10-K: Annual report pursuant to Section 13 and 15(d)
Published on February 23, 2017
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15 (d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2016
Or
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15 (d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number 033-80623
OncoGenex Pharmaceuticals, Inc.
(Exact name of the registrant as specified in its charter)
|
Delaware |
|
95-4343413 |
|
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
19820 North Creek Parkway, Bothell, Washington 98011
(Address of principal executive offices, including zip code)
(425) 686-1500
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of Each Class |
|
Name of Exchange on Which Registered |
|
Common Stock, par value $0.001 per share |
|
The NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such report), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|
|||
|
Non-accelerated filer |
|
☐ |
|
Smaller reporting company |
|
☒ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act.). Yes ☐ No ☒
As of June 30, 2016, the aggregate market value of the registrant’s Common Stock held by non-affiliates of the registrant was $29,650,858. As of Feb 23, 2017, 30,086,106 shares of the registrant’s Common Stock were outstanding.
OncoGenex Pharmaceuticals, Inc.
Table of Contents
|
|
|
|
|
|
|
ITEM 1. |
|
|
3 |
|
|
ITEM 1A. |
|
|
17 |
|
|
ITEM 1B. |
|
|
32 |
|
|
ITEM 2. |
|
|
32 |
|
|
ITEM 3. |
|
|
32 |
|
|
ITEM 4. |
|
|
32 |
|
|
|
|
|
||
|
|
|
|
|
|
|
ITEM 5. |
|
|
33 |
|
|
ITEM 6. |
|
|
35 |
|
|
ITEM 7. |
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
|
36 |
|
ITEM 7A. |
|
|
51 |
|
|
ITEM 8. |
|
|
52 |
|
|
ITEM 9. |
|
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
|
84 |
|
ITEM 9A. |
|
|
84 |
|
|
ITEM 9B. |
|
|
84 |
|
|
|
|
|
||
|
|
|
|
|
|
|
ITEM 10. |
|
|
85 |
|
|
ITEM 11. |
|
|
88 |
|
|
ITEM 12. |
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
|
104 |
|
ITEM 13. |
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
|
105 |
|
ITEM 14. |
|
|
106 |
|
|
|
|
|
||
|
|
|
|
|
|
|
ITEM 15. |
|
|
107 |
|
2
References in this Form 10-K to “OncoGenex Pharmaceuticals,” “OncoGenex,” the “Company,” “we,” “us” or “our” refer to OncoGenex Pharmaceuticals, Inc. and its wholly owned subsidiaries. The information in this Annual Report on Form 10-K contains certain forward-looking statements, including statements related to clinical trials, regulatory approvals, markets for our products, new product development, capital requirements and trends in our business that involve risks and uncertainties. Our actual results may differ materially from the results discussed in the forward-looking statements. Factors that might cause such a difference include those discussed in “Business,” “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” as well as those discussed elsewhere in this Annual Report on Form 10-K.
OVERVIEW OF OUR BUSINESS AND RECENT DEVELOPMENTS
We are a biopharmaceutical company that has been focused on the development of novel next generation cancer therapeutics. Our mission is to accelerate transformative therapies to improve the lives of people living with cancer and other serious diseases. Our product candidate apatorsen has a distinct mechanism of action and represents a unique opportunity for cancer drug development that we believe has the potential to improve treatment outcomes in a variety of cancers. Apatorsen is designed to block the production of heat shock protein 27, or Hsp27, a protein that promotes treatment resistance in cancer. In some clinical trials evaluating apatorsen, high serum Hsp27 levels appear to be a strong prognostic indicator for shorter survival outcomes. We currently do not intend to conduct additional pre-clinical or clinical studies with apatorsen and are seeking a collaboration partnership to fund and further develop this product candidate.
As a result of custirsen not meeting the primary endpoint of improving overall survival in three completed phase 3 trials, we have discontinued further development of custirsen and have begun to wind down all clinical trials and other activities related to this product candidate. In November 2016, we provided a notice of discontinuance to Ionis Pharmaceuticals, Inc. (formerly Isis Pharmaceuticals, Inc.), or Ionis, and a letter of termination to the University of British Columbia, or UBC, notifying those parties that we have discontinued development of custirsen, resulting in termination of all licensing agreements related to custirsen. In January 2017, we also discontinued further development of our pre-clinical product candidate, OGX-225. We provided a notice of discontinuance to Ionis, informing them that we have discontinued development of OGX-225 resulting in termination of the license agreement related to this product candidate. We intend to also terminate the UBC license agreement related to OGX-225, provided that Ionis does not exercise its reversion rights within 90 days of the notice of discontinuance. If Ionis exercises its reversion rights related to OGX-225, we believe Ionis will assume the rights and obligations under the UBC license agreement.
In February 2016, we committed to a plan to reduce operating expenses, which included a workforce reduction of 11 employees, representing approximately 27% of our employees prior to the reduction. We incurred approximately $0.4 million in expenses as a result of the workforce reduction, substantially all of which were severance costs.
In October and November 2016, we committed to a restructuring of an additional portion of our workforce in order to preserve our resources as we determined future strategic plans. As part of these restructurings, we eliminated 19 positions, representing approximately 68% of our workforce. We expect the restructurings to be substantially complete in the first quarter of 2017. In the fourth quarter of 2016, we incurred approximately $1.8 million in restructuring costs, substantially all of which related to severance costs, and an asset impairment charge of $0.2 million for manufacturing equipment.
On January 5, 2017, we and Achieve Life Science, Inc., or Achieve, a privately held specialty pharmaceutical company, entered into an Agreement and Plan of Merger and Reorganization, or the Merger Agreement, under which OncoGenex will acquire Achieve in an all-stock transaction. Upon completion of the Merger Agreement, Achieve's stockholders are expected to own 75% of the combined company's outstanding shares and our current equityholders are expected to own the remaining 25% of the combined company's outstanding shares. Following completion of the merger, OncoGenex Pharmaceuticals, Inc. will be renamed Achieve Life Sciences, Inc.
Pending Merger Agreement with Achieve
On January 5, 2017, we and Achieve entered into the Merger Agreement, pursuant to which Ash Acquisition Sub, Inc., a Delaware corporation and a wholly owned subsidiary of ours will merge with and into Achieve, or the First Merger, with Achieve becoming a wholly owned subsidiary of ours and the surviving company of the First Merger, or the Initial Surviving Corporation. Promptly following the First Merger, the Initial Surviving Corporation will merge with and into Ash Acquisition Sub 2, Inc., or Merger Sub 2, a Delaware corporation and a wholly owned subsidiary of ours, with Merger Sub 2 continuing as the surviving entity as a direct wholly owned subsidiary of ours. The two mergers taken together, are intended to qualify as a “reorganization” within the meaning of Section
3
368(a)(2)(D) of the Internal Revenue Code of 1986, as amended. The surviving company is expected to be renamed Achieve Life Sciences, Inc. and is referred to herein as the “combined company.” The Merger is expected to close mid-2017.
Subject to the terms and conditions of the Merger Agreement, at the closing of the First Merger, each outstanding share of Achieve common stock will be converted into the right to receive approximately 4,242.8904 shares of our common stock, subject to adjustment as provided in the Merger Agreement based on increases or decreases in Achieve’s fully-diluted capitalization, as well as the payment of cash in lieu of fractional shares. Immediately following the effective time of the merger, our equityholders are expected to own approximately 25% of the outstanding capital stock of the combined company on a fully diluted basis, and the Achieve stockholders are expected to own approximately 75% of the outstanding capital stock of the combined company on a fully diluted basis.
Consummation of the merger is subject to certain closing conditions, including, among other things, approval by the stockholders of us and Achieve. The Merger Agreement contains certain termination rights for both us and Achieve, and further provides that, upon termination of the Merger Agreement under specified circumstances, either party may be required to pay the other party a termination fee of $0.5 million. In addition, the Merger Agreement provides that if either party breaches certain covenants regarding alternative transactions to those contemplated by the Merger Agreement, the breaching party may be required to pay the other party a termination fee of $1.0 million. In connection with certain terminations of the Merger Agreement, either party may be required to pay the other party’s third party expenses up to $0.5 million.
At the effective time of the First Merger, our Board of Directors is expected to consist of seven members, three of whom will be designated by us and four of whom will be designated by Achieve. We are expected to designate Scott Cormack, Stewart Parker and Martin Mattingly. Achieve is expected to designate Richard Stewart, Anthony Clark and two other independent directors that have yet to be determined. Additionally, at the effective time of the First Merger, Rick Stewart, the current Chairman of Achieve, is expected to be the Chairman and Chief Executive Officer of the combined company; Anthony Clarke, the current Chief Scientific Officer of Achieve, is expected to be the Chief Scientific Officer of the combined company; and John Bencich, our Chief Financial Officer and Cindy Jacobs, our Chief Medical Officer, are expected to continue to serve the combined company in their respective roles.
In accordance with the terms of the Merger Agreement, (i) certain of our officers and directors, who collectively hold approximately 1.2 percent of the outstanding shares of our capital stock as of the close of business on January 4, 2017, have each entered into a support agreement with Achieve, or the OncoGenex Support Agreements, and (ii) certain officers, directors and stockholders of Achieve, who collectively hold approximately 78 percent of the outstanding shares of Achieve capital stock as of the close of business on January 4, 2017, have each entered into a support agreement with us, or the Achieve Support Agreements, and together with the OncoGenex Support Agreements, the Support Agreements. The Support Agreements include covenants as to the voting of such shares in favor of approving the transactions contemplated by the Merger Agreement and against actions that could adversely affect the consummation of the Merger.
The Support Agreements will terminate upon the earlier of the consummation of the First Merger or the termination of the Merger Agreement by its terms.
Concurrently and in connection with the execution of the Merger Agreement, (i) certain of our officers and directors, who collectively hold approximately 1.2 percent of the outstanding shares of our capital stock as of the close of business on January 4, 2017 and (ii) certain officers, directors and stockholders of Achieve, who collectively hold approximately 78 percent of the outstanding shares of Achieve capital stock as of the close of business on January 4, 2017, have each entered into lock-up agreements with us, pursuant to which, subject to certain exceptions, each stockholder will be subject to a 180-day, or the Lock-Up Period, lock-up on the sale of shares of our capital stock, which Lock-Up Period shall begin upon the consummation of the First Merger.
We expect to issue contingent value rights, or each, a CVR and collectively, the CVRs, to our existing stockholders prior to the completion of the First Merger. One CVR will be issued for each share of our common stock outstanding as of the record date for such issuance. Each CVR will be a non-transferable right to potentially receive certain cash, equity or other consideration received by the combined company in the event the combined company receives any such consideration during the five-year period after consummation of the First Merger as a result of the achievement of certain clinical milestones, regulatory milestones, sales-based milestones and/or up-front payment milestones relating to our product candidate apatorsen, or the Milestones, upon the terms and subject to the conditions set forth in a contingent value rights agreement to be entered into between us, Achieve and an as of yet unidentified third party, as rights agent, or the CVR Agreement. The aggregate consideration to be distributed to the holders of the CVRs, if any, will be equal to 80% of the consideration received by the combined company as a result of the achievement of the Milestones less certain agreed to offsets, as determined pursuant to the CVR Agreement. Under the CVR Agreement, for a period of six months beginning in February 2017, we will use certain defined efforts to enter into an agreement with a third party regarding the development and/or commercialization of apatorsen. At the expiration of this six-month period, if a third party has not entered into a term sheet for the development or commercialization of apatorsen, the combined company will no longer be contractually required to pursue an agreement regarding apatorsen and no consideration will be payable to the holders of CVRs.
4
Product Candidate Apatorsen Overview
Apatorsen is our product candidate that is designed to inhibit production of Hsp27, a cell-survival protein expressed in many types of cancers including bladder, prostate, breast, pancreatic and non-small cell lung cancer. Hsp27 expression is stress-induced, including by many anti-cancer therapies. Overexpression of Hsp27 is thought to be an important factor leading to the development of treatment resistance and is associated with metastasis and negative clinical outcomes in patients with various tumor types. In some clinical trials evaluating apatorsen, high serum Hsp27 levels at baseline, or at the start of treatment, appear to be a strong prognostic indicator for shorter survival outcomes.
In 2013, we initiated the ORCA (Ongoing Studies Evaluating Treatment Resistance in CAncer) program which encompasses six phase 2 clinical studies designed to evaluate whether treatment with apatorsen can lead to improved prognosis and treatment outcomes for cancer patients. Five of these trials have been completed and the remaining ongoing trial completed enrollment in 2016 with results expected in 2018. We currently do not intend to conduct additional pre-clinical or clinical studies with apatorsen and are seeking a collaboration partnership to fund and further develop this product candidate.
Custirsen
As a result of custirsen not meeting the primary endpoint of improving overall survival in three completed phase 3 trials, we have discontinued further development of custirsen and have begun to wind down all clinical trials and other activities related to this product candidate. In November 2016, we provided a notice of discontinuance to Ionis and a letter of termination to UBC, notifying those parties that we have discontinued development of custirsen, resulting in termination of all licensing agreements related to custirsen.
OGX-225
In January 2017, we discontinued further development of our pre-clinical product candidate, OGX-225. We provided a notice of discontinuance to Ionis, notifying them that we have discontinued development of OGX-225 resulting in termination of the license agreement related to this product candidate. We intend to also terminate the UBC license agreement related to OGX-225 provided that Ionis does not exercise its reversion rights within 90 days of the notice of discontinuance. If Ionis exercises its reversion rights related to OGX-225, we believe Ionis will assume the rights and obligations under the UBC license agreement.
Financial Overview
We have devoted substantially all of our resources to our clinical development programs.
In 2016, we recognized $5.1 million in collaboration revenue attributable to a collaboration agreement with Teva Pharmaceutical Industries Ltd., or Teva.
We incurred a loss for the year ended December 31, 2016 of $20.1 million and had an accumulated deficit at December 31, 2016 of $196.9 million and $27.5 million of total assets. We expect to continue to incur additional losses either in connection with completing the merger and continuing the research and development activities of the combined company’s product candidates or winding down our current product development activities.
To date, we have funded our operations primarily through the sale of our equity securities and payments received from Teva. We will not receive any further payments from Teva.
Based on our current expectations, we believe that our cash, cash equivalents, and short-term investments will be sufficient to fund our currently planned operations for at least the next 12 months.
OUR PRODUCT CANDIDATE - APATORSEN
Overview of Apatorsen
Apatorsen is our product candidate that is designed to inhibit production of Hsp27, a cell-survival protein expressed in many types of cancers including bladder, prostate, breast, pancreatic and non-small cell lung cancer. Hsp27 expression is stress-induced, including by many anti-cancer therapies. Overexpression of Hsp27 is thought to be an important factor leading to the development of treatment resistance and is associated with metastasis and negative clinical outcomes in patients with various tumor types. In some clinical trials evaluating apatorsen, high serum Hsp27 levels at baseline, or at the start of treatment, appear to be a strong prognostic indicator for shorter survival outcomes.
5
Apatorsen utilizes second-generation antisense drug chemistry and belongs to the drug class known as antisense therapeutics. We have collaborated with Ionis and selectively licensed technology from Ionis to combine Ionis’ second-generation antisense chemistry with our proprietary gene target sequences to create an inhibitor that is designed to down-regulate Hsp27. In contrast to first-generation antisense chemistry, second-generation antisense chemistry has improved target binding affinity, increased resistance to degradation and improved tissue distribution. These improvements result in slower clearance of the therapies from the body, which allow for less frequent dosing and thereby make treatment easier on patients at a lower associated cost.
A number of preclinical studies have shown that reducing Hsp27 production induces tumor cell death in prostate, non-small cell lung, bladder and pancreatic cancer cells. The studies also suggest that reducing Hsp27 production sensitizes prostate tumor cells to hormone ablation therapy. These preclinical studies have also shown that inhibiting the production of Hsp27 in human prostate, bladder, lung, breast, ovarian and pancreatic tumor cells sensitizes the cells to chemotherapy.
Hsp27 has been reported by others to function as an immunomodulatory protein by a number of mechanisms that include altering important membrane-expressed proteins on monocytes and immature dendritic cells; this alteration results in tumor-associated immune cells that are not functional in identifying and killing cancer cells. The induction of anti-inflammatory cytokines by Hsp27 may also play a role in down-regulating lymphocyte activation leading to additional unresponsive immune cells.
In 2013, we initiated the ORCA (Ongoing Studies Evaluating Treatment Resistance in CAncer) program which encompasses six phase 2 clinical studies designed to evaluate whether treatment with apatorsen can lead to improved prognosis and treatment outcomes for cancer patients. Five of these trials have been completed and the remaining ongoing trial completed enrollment in 2016 with results expected in 2018. We currently do not intend to conduct additional pre-clinical or clinical studies with apatorsen and are seeking a collaboration partnership to fund and further develop this product candidate.
Summary of Apatorsen Development Program
Ongoing Apatorsen Trial:
|
Cancer Indication and Trial |
|
Treatment Combination |
|
Status |
|
Advanced squamous NSCLC (Spruce-2) |
|
Gemcitabine and carboplatin with and without apatorsen (~ 90 patients) |
|
• Patient enrollment completed end of December 2016
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6
|
Cancer Indication |
|
Treatment Combination |
|
Status |
|
Advanced non-squamous NSCLC (Spruce) |
|
Carboplatin and pemetrexed with and without apatorsen (~155 patients) |
|
• Phase 2 top-line results on PFS reported in January 2016 and survival results reported below under the headings “Our Product Candidate - Apatorsen - Summary of Completed Apatorsen Clinical Trials” |
|
|
|
|
|
|
|
Metastatic bladder cancer (Borealis-2) |
|
Docetaxel with and without apatorsen (~ 200 patients); second-line chemotherapy |
|
• Final phase 2 data presented at 2017 GU ASCO |
|
|
|
|
|
|
|
Metastatic bladder cancer (Borealis-1) |
|
Gemcitabine and cisplatin with and without apatorsen (~ 180 patients); first-line chemotherapy |
|
• Final phase 2 data presented at 2015 ASCO Annual Meeting • Top-line data reported in December 2014 |
|
|
|
|
|
|
|
Metastatic pancreatic cancer (Rainier) |
|
Abraxane and gemcitabine with and without apatorsen (~ 130 patients) |
|
• Final phase 2 data presented at 2016 GI ASCO • Top-line data reported in September 2015 |
|
|
|
|
|
|
|
Castrate resistant prostate cancer (Pacific) |
|
Zytiga (abiraterone acetate) with and without apatorsen (~72 patients) |
|
• Final phase 2 data presented at 2017 GU ASCO. |
|
|
|
|
|
|
|
Solid tumors |
|
Apatorsen with and without chemotherapy |
|
• Final phase 1 data presented at 2010 ASCO Annual Meeting, |
|
|
|
|
|
|
|
Superficial and muscle invasive bladder Cancer (BL-01) |
|
Apatorsen as monotherapy (24 patients) |
|
• Preliminary phase 1 data presented at 2012 ASCO Genitourinary Cancers Symposium |
|
|
|
|
|
|
|
Castrate resistant prostate cancer (PR-01) |
|
Prednisone with and without apatorsen (74 patients) |
|
• Preliminary phase 2 data presented at 2012 ESMO Annual Meeting
|
Summary of Ongoing Trial
The Spruce-2™ Trial (formerly referred to as the Cedar Trial) is an investigator-sponsored, randomized phase 2 trial evaluating apatorsen plus gemcitabine and carboplatin therapy or gemcitabine and carboplatin therapy alone in patients with previously untreated advanced squamous NSCLC. Patients also continue weekly apatorsen infusions as maintenance treatment after chemotherapy until disease progression. The aim of the trial is to determine if adding apatorsen to gemcitabine and carboplatin therapy can extend progression free survival, or PFS, outcome. Additional analyses will include tumor response rates, overall survival, safety, and health-related quality of life, as well as to determine the effect of Hsp27 levels on clinical outcomes, explore potential biomarkers that may help predict response to treatment and survival outcomes in patients who were at increased risk for poor outcomes. The trial was initiated in July 2014 and completed enrollment in December 2016. During the conduct of the trial, two amendments were submitted: one that reduced the apatorsen dose to 400mg and the second that reduced patient enrollment to ~90 patients. The trial completed patient enrollment in December 2016 and results are expected in 2018. The trial is an investigator-sponsored trial being conducted and funded primarily by the UK National Cancer Research Network and the UK Experimental Cancer Medicine Network.
7
Summary of Completed Apatorsen Clinical Trials
The following is a summary of the preliminary or final results from completed apatorsen clinical trials.
Summary of Borealis-2 Results - The Randomized Phase 2 Clinical Trial in Patients with Metastatic Bladder Cancer who have disease progression following first-line platinum-based chemotherapy
Borealis-2 randomized 200 patients with metastatic bladder cancer whose disease had progressed following first-line platinum-based chemotherapy. Patients were randomized to receive docetaxel in combination with 600mg apatorsen or docetaxel alone. Patients could receive up to 10 cycles of docetaxel. Apatorsen maintenance could continue beyond docetaxel treatment until disease progression, toxicity, or study withdrawal. The primary endpoint analysis was a superiority test for overall survival, performed at a one-sided 0.10 significance level using a stratified log-rank test. Secondary endpoints included PFS, disease response and safety assessments. The Borealis-2 trial was an investigator-sponsored trial conducted by the Hoosier Cancer Research Network at 28 sites across the United States.
In October 2016, we announced that the trial met its primary endpoint of improving survival at the one-sided 0.10 significance level. Patients who received apatorsen treatment experienced a 20% reduction in risk of death, compared to patients receiving docetaxel alone (overall survival hazard ratio (HR)=0.80; 80% CI: 0.65-0.98; p=0.078). In February 2017, results were presented at the American Society of Clinical Oncology, or ASCO, 2017 Genitourinary Cancers Symposium. Apatorsen was well tolerated in combination with docetaxel. The reduction in risk of progression or death was also 20% for patients receiving apatorsen in combination with docetaxel, compared to docetaxel alone (PFS HR= 0.80; 80% CI: 0.64-1.01; p=0.107). Partial or complete responses occurred in 16.2% patients receiving apatorsen plus docetaxel compared to 10.9% patients receiving docetaxel alone with median response durations of 6.2 months versus 4.4 months, respectively. Overall for the study, higher baseline serum Hsp27 levels were significantly prognostic for indicating an almost 2-fold higher risk of death (HR= 1.96; p=0.0001). In an exploratory analysis on a subset of patients (20% of total) who completed at least two treatment cycles and had either a decrease in serum Hsp27 levels from baseline or had only a 20.5% increase in serum Hsp27 levels from baseline, the reduction in risk of death with apatorsen treatment was 71% (HR= 0.29: 80% CI: 0.18-0.48; interaction p=0.0727).
Summary of Borealis-1 Results - The Randomized Phase 2 Clinical Trial in Patients with Metastatic Bladder Cancer
Borealis-1 randomized 183 patients with documented metastatic or locally inoperable transitional cell carcinoma, or TCC, of the urinary tract who had not previously received chemotherapy for metastatic disease and were not candidates for potentially curative surgery or radiotherapy. Patients were randomized to receive standard chemotherapy (gemcitabine/cisplatin) in combination with apatorsen at two dose levels (600 mg and 1000 mg) or gemcitabine/cisplatin plus placebo. Patients received up to six cycles of weekly intravenous therapy. Patients received weekly apatorsen or placebo maintenance therapy until disease progression or other reason for withdrawal from protocol treatment if they had completed a minimum of four cycles of chemotherapy. The primary endpoint of the trial was overall survival. Secondary endpoints included PFS, disease response and safety assessments for the two doses of apatorsen. The trial was conducted by OncoGenex as a company-sponsored trial at 50 sites in the United States, Canada, and Europe.
In December 2014, we announced overall trial results that the addition of 600mg apatorsen to standard of care chemotherapy showed a 14% reduction in risk of death (HR = 0.86; 95% CI: 0.54-1.36; p=0.252) when compared to chemotherapy alone. Subsequent exploratory analyses showed a trend for improved survival in patients with baseline poor prognostic features treated with 600 mg apatorsen compared to placebo (HR=0.72; 95% CI: 0.35–1.45). In general for the study, higher baseline serum Hsp27 levels were significantly prognostic for indicating a 2-fold higher risk of death (HR= 2.01; p=0.0004). Further exploratory analysis of serum Hsp27 levels showed a trend towards survival benefit for the poor-prognosis patients in apatorsen 600 mg and 1000 mg arms who achieved lower overall (area-under-the-curve) serum Hsp27 levels during study treatment, compared to similar patients in the placebo arm (HR=0.45 and 0.62, respectively). Less benefit was believed to be observed in the 1000mg apatorsen arm due to increased adverse events leading to a higher rate of discontinuation of both apatorsen and chemotherapy. Apatorsen 600mg was well tolerated in combination with gemcitabine/cisplatin chemotherapy. These data were presented at the 2015 ASCO Annual Meeting.
Summary of Spruce Results - The Randomized Phase 2 Clinical Trial in Patients with Non-Small Cell Lung Cancer (NSCLC)
Spruce randomized 155 patients with previously untreated advanced non-squamous non-small cell lung cancer, or NSCLC. Patients were randomized to receive apatorsen in combination with carboplatin and pemetrexed therapy compared to carboplatin and pemetrexed therapy alone. Patients were to continue pemetrexed with weekly apatorsen or placebo infusions as maintenance treatment until disease progression if they completed a minimum of 3 cycles of chemotherapy treatment. The aim of the trial was to determine if adding apatorsen to carboplatin and pemetrexed therapy could extend PFS outcome. The study was an investigator-sponsored trial conducted by sites under the Sarah Cannon Research Institute.
8
In January 2016, the primary endpoint data for PFS was reported to have not reached the statistical significance required to demonstrate a benefit (PFS HR= 0.90; 80% CI 0.71-1.14; p=0.557). In the study, higher baseline serum Hsp27 levels were found to be significantly prognostic for indicating an almost 2-fold higher risk of death (HR= 1.98; p=0.0034). A potential benefit was observed in a subgroup of patients with high baseline serum Hsp27 status (~10% of total) when treated with apatorsen (PFS HR= 0.462; 80% CI: 0.193- 1.106). Study follow up with survival results was completed at the end of 2016. The addition of apatorsen to carboplatin and pemetrexed therapy did not demonstrate an overall survival benefit in the study (HR= 1.067; 80% CI: 0.838-1.359). PFS results were presented at ASCO 2016. The study investigators concluded that apatorsen and pemetrexed/carboplatin therapy was well tolerated and showed promising PFS results in the treatment of patients with non-squamous NSCLC who have Hsp27 high status and thus warranted further study in this population. We do not intend to pursue additional trials in non-squamous NSCLC at this time.
Summary of Rainier Results - The Randomized Phase 2 Clinical Trial in Patients with Untreated Metastatic Pancreatic Cancer
Rainier randomized 132 patients with previously untreated metastatic pancreatic cancer. Patients were randomized to receive apatorsen in combination with ABRAXANE® (paclitaxel protein-bound particles for injectable suspension) (albumin-bound) and gemcitabine compared to ABRAXANE and gemcitabine alone. Patients were to receive up to six cycles of weekly intravenous therapy. The aim of the trial was to determine if adding apatorsen to ABRAXANE and gemcitabine could extend overall survival. The study was an investigator-sponsored trial conducted by sites under the Sarah Cannon Research Institute.
In September 2015, we announced that the primary survival endpoint did not show improved survival for patients receiving apatorsen plus ABRAXANE and gemcitabine when compared to ABRAXANE and gemcitabine alone (HR= 1.098; 95% CI 0.759-1.590). Similarly there was no improvement in PFS (PFS HR=1.020; 95% CI 0.806-1.290). The study did show that higher baseline serum Hsp27 levels were significantly prognostic for indicating a 1.8-fold higher risk of death (HR= 1.84; p=0.0041). A potential benefit was observed in a subgroup of patients with high baseline serum Hsp27 status (14% of total) when treated with apatorsen (PFS HR= 0.381; 95% CI 0.120-1.208 and survival HR= 0.587; 95% CI 0.195-1.770). The study was presented at the Gastrointestinal, or GI, Cancers Symposium meeting in January 2016. The study investigators concluded that apatorsen and ABRAXANE/gemcitabine was well tolerated and that the promising results in pancreatic cancer patients with high baseline Hsp27 status warrant further study of apatorsen in this population. We do not intend to pursue additional trials in pancreatic cancer at this time.
Summary of Pacific Results - The Randomized Phase 2 Clinical Trial in Patients with metastatic CRPC
Pacific randomized 72 patients who were experiencing a rising PSA while receiving Zytiga® (abiraterone acetate). The aim of the trial was to determine if adding apatorsen to Zytiga treatment could reverse or delay treatment resistance by evaluating the PFS rate at a milestone Day 60 assessment. The primary endpoint was the proportion of patients who were progression free (clinical and radiologic) at study day 60. Other secondary endpoints were PSA and objective responses, time to disease progression, circulating tumor cells, or CTCs, and Hsp27 levels. The Pacific trial was an investigator-sponsored trial conducted by the Hoosier Cancer Research Network at sites in Canada and the United States.
In February 2017, results were presented at the ASCO 2017 Genitourinary Cancers Symposium. Apatorsen was well tolerated in combination with Zytiga with the median treatment duration of 106 days for apatorsen plus Zytiga compared to 75 days for continuing Zytiga alone. The proportion of patients who were progression free at Day 60 was 33% when apatorsen was added to Zytiga, compared to 17% with Zytiga alone (p=0.17). The median time of PFS was 8.6 weeks for apatorsen treatment, compared to 7.9 weeks for Zytiga. A 50% or greater decline in PSA levels was seen in 6% of patients when apatorsen was added to Zytiga compared to 3% with continuing Zytiga alone. Stable disease or partial response was seen in 20% of patients when apatorsen was added to Zytiga vs 14% with Zytiga alone. For patients with ≥5 CTCs at baseline, 22% vs 11% of patients had a CTC reduction to less than 5 CTCs when apatorsen was added to Zytiga vs Zytiga alone, respectively.
Summary of Results of Apatorsen Randomized Phase 2 Clinical Trial in Patients with CRPC
This randomized, controlled phase 2 trial completed enrollment of 74 patients who had minimally symptomatic or asymptomatic advanced prostate cancer and who have not yet received chemotherapy. The trial was designed to determine the potential benefit of apatorsen by assessing the number of patients without disease progression at 12 weeks post-study treatment with or without apatorsen. Preliminary study results presented at ESMO in September 2012 showed a higher number of patients without disease progression at 12 weeks and greater declines in PSA and CTCs in patients receiving apatorsen plus prednisone treatment compared to those receiving prednisone alone. Apatorsen was well tolerated in combination with prednisone.
9
Summary of Results of Apatorsen Phase 1 Clinical Trial in Patients with Superficial Bladder Cancer
This investigator-sponsored phase 1 trial was designed to determine the effects of apatorsen on Hsp27 expression and tumor response rates when administered into the bladder using intravesical instillation. In addition, the trial measured the direct effect of delivering apatorsen by intravesical instillation on expression of Hsp27 in bladder tumor cells. This clinical trial was primarily funded by the National Cancer Institute of Canada.
Preliminary results from this trial were presented at the ASCO 2012 Genitourinary Cancers Symposium in February 2012 and demonstrated a trend towards decreased levels of Hsp27 and increased tumor cell death rates after intravesical treatment with apatorsen. In the apatorsen treated patients who experienced a complete pathologic response, the absence of residual disease made it difficult to fully assess the effect of apatorsen on Hsp27 expression. Therefore, the analysis was based mainly on the remaining patients who had evaluable tumor tissue. Results showed that eight of 24 patients (33%) had no pathologic evidence of disease.
Summary of Results of Apatorsen Phase 1 Clinical Trial in Patients with Solid Tumors
Apatorsen has been evaluated in a phase 1 trial in patients with breast, prostate, ovarian, or NSCLC who have failed potentially curative treatments or for whom a curative treatment does not exist. Final results of this phase 1 trial were presented in an oral presentation at the ASCO 2010 annual meeting. The phase 1 trial evaluated 42 patients treated with apatorsen as a single agent and 22 patients treated with apatorsen in combination with docetaxel who had failed up to six prior chemotherapy treatments. Apatorsen as a single agent administered weekly was evaluated at doses from 200 mg up to 1000 mg in five cohorts of approximately six patients per cohort. Two further cohorts evaluated apatorsen at the 800 and 1000 mg doses combined with docetaxel. Patients could receive up to 10 21-day cycles.
Most adverse events were mild (grade 1 or 2) and mainly occurred during the three “loading doses” given over nine days prior to weekly dosing. The most frequently reported adverse events in the apatorsen monotherapy arms were infusion-related reactions and chills. The most frequently reported adverse events in the apatorsen plus docetaxel arms were infusion-related reactions, chills, fatigue, diarrhea, pruritus (itching), nausea and back pain. The incidence of laboratory toxicity was determined based on laboratory data. The majority of laboratory toxicities were Grade 1 or Grade 2. Serious adverse events were reported for approximately half the patients. The most common events were disease progression and dyspnea (shortness of breath), reported for five subjects each, and febrile neutropenia, reported for four subjects. Increased blood creatinine (a test of kidney function) and hydronephrosis (obstruction of the urine flow from the kidney due to tumor blockage) were reported for two subjects each. All remaining serious adverse events were reported for one subject each.
Thirty patients had baseline and at least one post-baseline assessment of measurable disease. A total of eight of 30 patients (27%) had a decrease in measurable disease from baseline of at least 15%. For patients treated with monotherapy, three patients had tumor reductions and for patients treated with combined therapy with docetaxel, five patients had tumor reductions.
Thirty-three of 36 patients with prostate cancer had at least one post-baseline PSA. Three of 21 in the monotherapy cohorts had reductions in PSA greater than or equal to 30% as did six of 12 in the combination therapy cohorts. Six of seven patients with ovarian cancer had both baseline and post-baseline CA-125 (an ovarian tumor marker) measurements. All were treated with monotherapy. Three patients had a reduction of CA-125.
Decreases in both total CTCs and Hsp27+CTCs were observed. Hsp27+CTCs were decreased in 71% of evaluable patients.
In approximately 35% of patients, serum Hsp27 protein levels were decreased by 30% or greater over a time period of at least six weeks.
OVERVIEW OF MARKET AND TREATMENT
In North America, cancer has recently surpassed heart disease as the leading cause of death in the United States. The American Cancer Society estimates that approximately 1.7 million new cancer cases are expected to be diagnosed in 2017. Cancer is the second most common cause of death in the United States, accounting for nearly 1 of every 4 deaths. Approximately 600,000 Americans are expected to die of cancer in 2017.
Typically, cancer treatments are given sequentially and can include hormone therapy, surgery, radiation therapy, immunotherapy and chemotherapy. Although a particular therapy may initially be effective, tumor cells often react to therapeutic treatment by increasing the production of proteins that afford them a survival advantage, enabling them to become resistant to therapy, multiply, and spread to additional organs. As a result, many patients progress through multiple different therapies and ultimately die from the disease.
10
LICENSE AND COLLABORATION AGREEMENTS
Ionis Pharmaceuticals, Inc.
Apatorsen
In January 2005, we entered into a collaboration and license agreement with Ionis to jointly identify antisense compounds designed to inhibit the production of proteins encoded by specified gene targets. We are solely responsible for all product development activities for antisense compounds under this collaboration. This relationship provides us with access to Ionis’ proprietary position in second generation antisense chemistry for use in specified products. We were permitted to designate up to two collaboration gene targets for collaborative research, development and commercialization. In April, 2005, Hsp27 was confirmed as a collaboration gene target, and we and Ionis jointly designed and screened antisense compounds for this gene target. Our right to designate a second collaboration gene target expired on January 5, 2007.
Under the terms of the agreement, in the event that we abandon apatorsen, Ionis may elect to unilaterally continue development of apatorsen, in which case we must provide Ionis with a worldwide license or sublicense (as the case may be) of our relevant technology solely to develop and commercialize apatorsen in exchange for a royalty on Ionis’ sales of apatorsen.
Under the terms of the agreement, we may be obligated to make certain milestone payments to Ionis contingent upon the occurrence of certain clinical development and regulatory events related to apatorsen. We are also obligated to pay to Ionis certain milestone payments, as well as certain low to mid-single digit royalties on net sales for apatorsen, with the amount of royalties depending on whether third-party royalty payments are owed. We paid Ionis USD$0.8 million in 2010 upon the initiation of a phase 2 clinical trial of apatorsen in patients with CRPC. We did not make any royalty payments to Ionis under the terms of the agreement in 2016.
We have agreed to indemnify Ionis and certain persons affiliated with Ionis against liabilities caused by us and our licensees’ and sublicensees’ gross negligence or willful misconduct, our material breach of the collaboration and license agreement, and the manufacture, use, handling, storage, sale or other disposition of apatorsen that is sold by us or our affiliates, agents or sublicensees.
The term of the collaboration and license agreement will continue for each product until the later of 10 years after the date of the first commercial sale of apatorsen and the expiration of the last to expire of any patents required to be licensed in order to use or sell apatorsen, unless we abandon apatorsen and Ionis does not elect to unilaterally continue development of apatorsen.
Custirsen
In November 2016, we provided a notice of discontinuance to Ionis notifying them that we have discontinued development of custirsen, resulting in termination of all licensing agreements related to custirsen. We believe that all financial obligations, other than continuing mutual indemnification obligations and our requirement to pay for out-of-pocket patent expenses incurred up to the date of termination and for abandoning the custirsen patents and patent applications, under all agreements with Ionis, including the Ionis settlement agreement, are no longer owed and no further payments are due.
OGX-225
In January 2017, we discontinued further development of OGX-225. We provided a notice of discontinuance to Ionis, notifying them that we have discontinued development of OGX-225 resulting in termination of the license agreement related to this product candidate. We intend to also terminate the UBC license agreement related to OGX-225 provided that Ionis does not exercise its reversion rights within 90 days of the notice of discontinuance. If Ionis exercises its reversion rights related to OGX-225, we believe Ionis will assume the rights and obligations under the UBC license agreement.
University of British Columbia
Apatorsen
Under a license agreement entered into in April 2005, as amended, UBC granted to us an exclusive, worldwide license to commercialize its existing intellectual property and any improvements related to Hsp27. This technology, combined with Ionis’ second-generation antisense chemistry, is our product candidate apatorsen. In connection with entering into the license agreement, we issued to UBC shares that were exchanged in the Arrangement for 6,533 shares of our common stock. We also agreed to pay UBC low single digit royalties on the revenue from sales of apatorsen, which royalty rate may be reduced in the event that we must pay additional royalties under patent licenses entered into with third parties in order to manufacture, use or sell apatorsen. We may be obligated to make milestone payments to UBC contingent upon the occurrence of certain clinical development and regulatory events
11
related to apatorsen. We are obligated to pay UBC CAD$2,000 in annual maintenance fees. We paid UBC CAD$0.1 million in 2010 in relation to the initiation of a phase 2 trial of apatorsen in patients with CRPC. The occurrence and receipt of upfront and milestone payments and the generation of royalty revenue are uncertain. We did not make any royalty payments to UBC under the terms of the agreement in 2016.
Subject to certain exceptions, we agreed to use our commercially reasonable efforts to (i) develop and exploit the licensed technology and any improvements and (ii) promote, market and sell any resulting products. We are permitted to sublicense the technology, subject to certain consent and other requirements. We direct patent prosecution and are responsible for all fees and costs related to the preparation, filing, prosecution and maintenance of the patent rights underlying the license agreement. We indemnify UBC and certain of UBC’s affiliates against liability arising out of the exercise of any rights granted pursuant to the agreement. The term of the agreement will expire on the later of 20 years from its effective date and the expiration of the last patent licensed under the agreement. Depending on the outcome of the pending patent applications in the licensed patent family, and subject to any applicable patent term extensions, a patent issuing from this family would expire in all jurisdictions by 2023. We may also file additional patent applications related to Hsp27 that could potentially extend the expiration date of the last to expire patent in this area.
Custirsen
In November 2016, we provided a letter of termination to UBC notifying them that we have discontinued development of custirsen, resulting in termination of all licensing agreements related to custirsen. We believe that all financial obligations, other than continuing mutual indemnification obligations and our requirement to pay for out-of-pocket patent expenses incurred up to the date of termination and for abandoning the custirsen patents and patent applications, under all agreements with UBC, are no longer owed and no further payments are due.
OGX-225
In January 2017, we discontinued further development of OGX-225. We provided a notice of discontinuance to Ionis, notifying them that we have discontinued development of OGX-225 resulting in termination of the license agreement related to this product candidate. We intend to also terminate the UBC license agreement related to OGX-225 provided that Ionis does not exercise its reversion rights within 90 days of the notice of discontinuance. If Ionis exercises its reversion rights related to OGX-225, we believe Ionis will assume the rights and obligations under the UBC license agreement.
Summary of Milestone Obligations by Product Candidate
The following table sets forth the milestones that we may be required to pay to third parties under the license and collaboration agreements described above. As described above, we will also be required to pay certain revenue-based royalties with respect to our product candidate.
|
Milestone Obligations to Third Parties |
|
Amount Payable |
|
Apatorsen |
|
Up to $4,808,000 (1)(2)(3) |
|
OGX-225 |
|
Up to $4,132,000 (3)(4) |
|
(1) |
Additional milestone payments may be required for product approvals outside the field of oncology. |
|
(2) |
Payable in connection with initiating certain clinical trials and obtaining certain market approvals. |
|
(3) |
Certain milestone payments are payable in Canadian dollars, which are translated based on the December 31, 2016 exchange rate of US$1.00 = CAD$1.34551 and rounded to the nearest $1,000. |
|
(4) |
Product candidate in process of being discontinued. |
GOVERNMENT REGULATIONS
Drug Approval Process
Regulation by government authorities in the United States and other countries is a significant factor in our ongoing research and development activities and in the production and marketing of our products. In order to undertake clinical trials and to produce and market products for human use, mandatory procedures and safety standards established by the FDA in the United States and by comparable agencies in other countries must be followed.
12
The standard process before a pharmaceutical agent may be marketed includes the following steps:
|
|
• |
preclinical studies, including laboratory evaluation and animal studies to test for initial safety and efficacy; |
|
|
• |
submission to national health authorities of an IND or Clinical Trials Application, or CTA, or equivalent dossier, which must be accepted by each national health authority before human clinical trials may commence in that country; |
|
|
• |
adequate and well-controlled clinical trials to establish the safety and efficacy of the drug in its intended population and use(s); |
|
|
• |
submission to appropriate national and/or regional regulatory health authorities of a New Drug Application, or NDA, or equivalent marketing authorization application, which application is not automatically accepted for review; and |
|
|
• |
approval by appropriate regulatory health authorities of the marketing authorization application prior to any commercial sale or shipment of the drug in each country or jurisdiction. |
As part of the regulatory health authority approval for each product, the drug-manufacturing establishment is subject to inspection by the FDA and must comply with current Good Manufacturing Practices, or cGMP, requirements applicable to the production of pharmaceutical drug products. The facilities, procedures and operations of manufacturers must be determined to be adequate by the FDA before product approval.
Preclinical studies include laboratory evaluation of the active drug substance and its formulation in animals to assess the potential safety and efficacy of the drug and its formulation. Prior to initiating the first clinical testing of a new drug product candidate, the results of the preclinical studies are submitted to regulatory health authorities as part of an IND or CTA, and must be accepted before the proposed clinical trial(s) can begin.
Clinical trials for cancer therapeutics involve the administration of the investigational drug to patients with a defined disease state, under the supervision of a qualified principal investigator.
Clinical trials are conducted in accordance with protocols that detail the parameters to be used to monitor safety and efficacy. Each protocol is submitted to regulatory health authorities as part of the IND or CTA in each country where clinical trials are to be conducted. Each clinical trial is approved and monitored by independent Institutional Review Boards, or IRB, or Ethics Committees who consider ethical factors, informed consent documents, the safety of human subjects and the possible liability of the institutions conducting a clinical trial. The IRB or Ethics Committee may require changes in the clinical trials protocol, which may delay initiation or completion of the trial.
Clinical trials typically are conducted in three sequential phases, although the phases may overlap. In phase 1, the initial introduction of the drug to humans, the drug is tested for safety and clinical pharmacology. Phase 2 trials involve more detailed evaluation of the safety and efficacy of the drug in patients with a defined disease. Phase 3 trials consist of large-scale evaluations of safety and efficacy of the investigational drug compared to accepted standard therapy in a defined disease.
The process of completing clinical testing and obtaining regulatory approval for a new product takes a number of years and requires the expenditure of substantial resources. The FDA, or another regulatory authority, may not grant approval on a timely basis, if at all. We may encounter difficulties in securing regulatory approval or unanticipated costs, which may delay or preclude the commercialization, if any, of apatorsen or future product candidates. For instance, regulatory authorities may conclude that the data submitted in a marketing authorization application, such as a NDA, are not adequate to support approval of a pharmaceutical agent and may require further clinical and preclinical testing, re-submission of the application, and further review. Even after initial approval has been obtained, an indication may be limited or conditioned on the provision of further studies to support an approved indication, and further studies will be required to gain approval for the use of a product for clinical indications other than those for which the product was approved initially. Also, regulatory authorities require post-marketing surveillance programs to monitor the drug product’s side effects.
Marketing of pharmaceutical products outside of the United States is subject to regulatory requirements that vary from country to country. In the European Union, the general trend has been towards coordination of common standards for clinical testing of new drug products. Centralized approval in the European Union is coordinated through the EMA.
The level of regulation outside the United States and the European Union varies widely. The time required to obtain regulatory approval from regulatory agencies in each country may be longer or shorter than that required for FDA or EMA approval. In addition, in certain markets, reimbursement is subject to governmentally mandated prices.
13
Our strategy is to outsource certain product development activities and have established contract research agreements for, preclinical, clinical, manufacturing and some data management services. We choose which business or institution to use for these services based on their expertise, capacity and reputation and the cost of the service.
We also provide or have provided quantities of our product candidates to academic research institutions to investigate the mechanism of action and evaluate novel combinations of product candidates with other cancer therapies in various cancer indications. These collaborations expand our research activities for our product candidates with modest contribution from us.
RESEARCH AND DEVELOPMENT EXPENDITURES
For the years ended December 31, 2016, 2015 and 2014, our expenditures for research and development activities were $14.8 million, $25.1 million and $46.2 million, respectively. Such research and development expenses primarily related to the advancement of our product candidates custirsen and apatorsen.
MANUFACTURING
We do not own facilities for the manufacture of materials for clinical or commercial use. We rely and expect to continue to rely on contract manufacturers to manufacture our product candidate in accordance with cGMP, for use in clinical trials, as well as for process development as required.
To date, all active pharmaceutical ingredient, or API, and drug product for apatorsen has been manufactured by third parties on a purchase order basis, under cGMP.
INTELLECTUAL PROPERTY
Our success depends in part on our ability and that of our collaborators to obtain and maintain proprietary protection for our product candidate, technology, and know-how, to prevent others from infringing on the proprietary rights of our product candidate, and to operate without infringing on the proprietary rights of others.
Patents
We have a license from UBC and Ionis to use, make, have made, or make improvements upon apatorsen. In addition, we have a pending family of applications on an apatorsen formulation.
We have been granted non-exclusive rights to all intellectual property owned, licensed or otherwise controlled by Ionis as of the date of our agreements with Ionis that relate to second-generation antisense chemistry and that are required for apatorsen. Ionis is generally restricted from engaging in research, development and commercialization of antisense compounds related to Hsp27, other than as provided in the collaboration and license agreement related to each target. Ionis directs patent prosecution and is responsible for all fees and costs related to the preparation, filing, prosecution and maintenance of these patent rights, which extend to numerous jurisdictions throughout the world. Individual patents have terms of protection depending on the laws of the countries in which the applications are made.
We direct patent prosecution, and are responsible for all fees and costs related to the preparation, filing, prosecution and maintenance of the patent rights for intellectual property under license from UBC covering apatorsen. We file patent applications for this intellectual property in the United States, Canada, Europe (through the European Patent Office), Japan and other jurisdictions.
Composition of matter patents covering apatorsen have been issued in the United States and certain other jurisdictions. Additional patent applications covering all of these products, as well as other technologies, are pending in the United States and certain other countries.
Generally, patents issued in the United States are effective for 20 years from the earliest non-provisional filing date, if the application from which the patent issues was filed on or after June 8, 1995 (otherwise the term is the longer of 17 years from the issue date and 20 years from the earliest non-provisional filing date). The duration of patent terms for non-U.S. patents is typically 20 years from the earliest corresponding national or international filing date. Our licensed UBC patent estate related to apatorsen, based on those patents that exist now, will expire in 2023, which does not include extensions that may be available or patent applications that are currently pending. Patent term extensions, specifically to make up for regulatory delays, are available in the United States, Europe and Japan.
14
Although we believe that some or all patents related to apatorsen will meet the criteria for patent term extensions, we can provide no assurance that we will obtain such extensions.
We also rely on unpatented trade secrets, proprietary know-how and continuing technological innovation, which we seek to protect, in part, by confidentiality agreements with our corporate partners, collaborators, employees and consultants in our drug development research. We can provide no assurance that these agreements will not be breached, that we will have adequate remedies for any breach, or that our trade secrets or know-how will not otherwise become known or be independently discovered by competitors. Further, we can provide no assurance that we will be able to protect our trade secrets or that others will not independently develop substantially equivalent proprietary information and techniques.
Trademarks
We own several trademarks registered in the United States, including word marks ONCOGENEX™, ORCA™, Spruce™, and design marks ORCA, Pacific, Borealis-1, Borealis-2, and the helical totem element that accompanies the clinical trial trademarked identifiers. In Canada, we have corresponding trademark registrations.
We can provide no assurance that our registered or unregistered trademarks or trade names will not infringe upon third-party rights or will be acceptable to regulatory agencies.
COMPETITION
The life sciences industry is highly competitive, and we face significant competition from many pharmaceutical, biopharmaceutical and biotechnology companies that are researching and marketing products designed to address cancer indications for which we are currently developing products or for which we may develop products in the future. We are aware of several other companies which are developing therapeutics that seek to promote tumor cell death. Several therapies have been recently approved by the FDA, and we expect more to be approved in the future.
Many oncology drugs in clinical trials are being developed for bladder, lung and prostate cancers. Certain of these drugs are designed, like apatorsen, to interfere with mechanisms potentially involved with treatment resistance. If new drugs are approved for sale for the indications that we are evaluating apatorsen, whether or not they are targeting mechanisms of treatment resistance, in advance of apatorsen or even after its commercialization, the market’s interest in apatorsen may be reduced or eliminated. We are aware of several other companies developing therapeutic products, whether antisense or otherwise, which seek to promote tumor cell death by inhibiting proteins believed to promote cell survival. Our competitors may seek to identify gene sequences, protein targets or antisense chemistry different from ours, and outside the scope of our intellectual property protection, to develop antisense therapeutics that serve the same function as apatorsen. Our competitors may also seek to use mechanisms other than antisense to inhibit the proteins that apatorsen is designed to inhibit.
Apatorsen has been evaluated in bladder, prostate, pancreas and lung cancer indications. Substantial advancements in the treatment of each of these cancers has occurred in the past several years and new products from our competitors have been approved for marketing on the basis of showing a survival advantage. Many of our existing and potential competitors have substantially greater financial resources and expertise than we do in manufacturing and developing products, conducting clinical trials, obtaining regulatory approvals and marketing. These entities also compete with us in recruiting and retaining qualified scientific and management personnel, as well as in acquiring products and technologies complementary to our programs. Standard treatments vary considerably by cancer indication, and new drugs may be more effective in treating one cancer indication than another. In addition, cancer is a difficult disease to treat and it is likely that no one therapeutic will replace all other therapies in any particular indication. Therapeutic strategies for treating cancer are increasingly focused on combining a number of drugs in order to yield the best results. Since apatorsen can potentially be used in multiple cancer indications and target the tumors’ adaptive survival mechanisms, it may potentially be synergistic with many new and currently marketed therapies. The ability for apatorsen to be developed and compete successfully will depend largely on our ability to find a collaboration partner willing to fund the future development and commercialization of this product candidate and for that collaboration partner, if any, to:
|
|
• |
maintain or establish development programs in combination with new agents that may replace or diminish the markets for which we are currently developing apatorsen; |
|
|
• |
establish that apatorsen is well tolerated and result in a clinical benefit when administered to cancer patients; |
|
|
• |
establish that apatorsen addresses significant unmet needs for patients, resulting in prioritization of apatorsen over other treatment options; |
|
|
• |
advance the development of apatorsen, including the enrollment of patients for our clinical trials; |
15
|
|
• |
gain regulatory approval for apatorsen in its first indication as well as expand into additional indications; |
|
|
• |
commercialize apatorsen successfully, which includes convincing physicians, insurers and other third-party payors of the advantages of apatorsen over current therapies, when and if they have advantages; and |
|
|
• |
obtain intellectual property protection and protect the exclusivity for apatorsen, when and if we have any. |
EMPLOYEES
As of December 31, 2016, we had a total of 17 employees, of whom 10 were engaged in research and development functions, including clinical development, regulatory affairs and manufacturing, and 7 were engaged in general and administrative functions, including accounting and finance, administration, and corporate communications.
All of our employees have entered into non-disclosure agreements regarding our intellectual property, trade secrets and other confidential information. None of our employees are represented by a labor union or covered by a collective bargaining agreement, nor have we experienced any work stoppages. We believe that we maintain satisfactory relations with our employees.
From time to time, we also use outside consultants to provide advice on our clinical development plans, research programs, administration and potential acquisitions of new technologies.
FINANCIAL INFORMATION
We manage our operations and allocate resources as a single reporting segment. Financial information regarding our operations, assets and liabilities, including our total revenue and net loss for the years ended December 31, 2016, 2015 and 2014 and our total assets as of December 31, 2016 and 2015, is included in our Consolidated Financial Statements in Part II, Item 8 of this Annual Report on Form 10-K.
COMPANY INFORMATION
We were incorporated in California in October 1991 and subsequently reorganized as a Delaware corporation in March 1995. Our principal executive offices are located at 19820 North Creek Parkway, Bothell Washington 98011, and our telephone number is (425) 686-1500.
In August 2008, our company, then named Sonus Pharmaceuticals, Inc., completed its acquisition, or the Arrangement, of OncoGenex Technologies, a Canadian corporation, as contemplated by the Arrangement Agreement between the companies. We then changed our name to OncoGenex Pharmaceuticals, Inc. As a result of the Arrangement, OncoGenex Technologies became our wholly owned subsidiary. OncoGenex Technologies was incorporated under the federal laws of Canada in May 2000. OncoGenex, Inc., a former subsidiary of OncoGenex Technologies, was incorporated under the laws of Washington in August 2005 and was dissolved pursuant to the Articles of Dissolution filed on July 1, 2009. As used in this Annual Report on Form 10-K, the term “Sonus” refers to our business prior to August 21, 2008.
AVAILABLE INFORMATION
We maintain a website at http://www.oncogenex.com. The information contained on or accessible through our website is not part of this Annual Report on Form 10-K. Our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended, or Exchange Act, are available free of charge on our website as soon as reasonably practicable after we electronically file such reports with, or furnish those reports to, the SEC. Any information we filed with the SEC may be accessed and copied at the SEC’s Public Reference Room at 100 F Street NE, Washington, DC 20549. Information may be obtained by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC at http://www.sec.gov.
16
Risks Related to Our Business
Investing in our common stock involves a high degree of risk. You should consider carefully the risks and uncertainties described below, together with all of the other information contained in this Annual Report on Form 10-K and in the other periodic and current reports and other documents we file with the Securities and Exchange Commission, before deciding to invest in our common stock. If any of the following risks materialize, our business, financial condition, results of operation and future prospects will likely be materially and adversely affected. In that event, the market price of our common stock could decline and you could lose all or part of your investment.
Risks Related to Our Pending merger
There is no assurance that the proposed merger between us and Achieve will be completed in a timely manner or at all. If the merger with Achieve is not consummated, our business could suffer materially and our stock price could decline.
The consummation of the proposed merger between us and Achieve is subject to a number of closing conditions, including the approval by our stockholders and other customary closing conditions. The parties are targeting a closing of the transaction in mid-2017. However, there can be no assurance that the proposed merger will be consummated on the desired timeframe, or at all.
If the proposed merger between us and Achieve is not consummated, we may be subject to a number of material risks, and our business and stock price could be adversely affected, as follows:
|
|
• |
|
we have incurred and expect to continue to incur significant expenses related to the proposed merger with Achieve even if the merger is not consummated; |
|
|
• |
|
we could be obligated to pay Achieve up to a $1.0 million termination fee and/or up to $0.5 million in merger related expenses in connection with the termination of the merger agreement, depending on the reason for the termination; |
|
|
• |
|
the market price of our common stock may decline to the extent that the current market price reflects a market assumption that the proposed merger will be completed; and |
|
|
• |
|
we may not be able to pursue an alternate merger transaction if the proposed merger with Achieve is not completed. |
If the merger is not completed, our board of directors may decide to pursue a dissolution and liquidation of the company. In such an event, the amount of cash available for distribution to our stockholders will depend heavily on the timing of such liquidation as well as the amount of cash that will need to be reserved for commitments and contingent liabilities.
There can be no assurance that the merger will be completed. If the merger is not completed, our board of directors may decide to pursue a dissolution and liquidation of the company. In such an event, the amount of cash available for distribution to our stockholders will depend heavily on the timing of such decision, as with the passage of time the amount of cash available for distribution will be reduced as we continue to fund our operations. In addition, if our board of directors was to approve and recommend, and our stockholders were to approve, a dissolution and liquidation of the company, we would be required under Delaware corporate law to pay out outstanding obligations, as well as to make reasonable provision for contingent and unknown obligations, prior to making any distributions in liquidation to our stockholders. Our commitments and contingent liabilities may include severance obligations, regulatory and clinical obligations remaining under our clinical trials, fees and expenses related to the merger and non-cancelable lease obligations. As a result of this requirement, a portion of our assets may need to be reserved pending the resolution of such obligations. In addition, we may be subject to litigation or other claims related to a dissolution and liquidation. If a dissolution and liquidation were pursued, our board of directors, in consultation with its advisors, would need to evaluate these matters and make a determination about a reasonable amount to reserve. Accordingly, holders of our common stock could lose all or a significant portion of their investment in the event of a liquidation, dissolution or winding up of our company.
The issuance of shares of our common stock to Achieve stockholders in the pending merger will dilute substantially the voting power of our current stockholders.
If the pending merger is completed, each outstanding share of Achieve common stock will be converted into the right to receive approximately 4,242.8904 shares of our common stock, subject to certain adjustments. Immediately following the merger, our equityholders are expected to own approximately 25% of the outstanding capital stock of the combined company on a fully diluted basis, and the Achieve stockholders are expected to own approximately 75% of the outstanding capital stock of the combined company on a fully diluted basis. Accordingly, the issuance of shares of our common stock to Achieve stockholders in the merger will
17
reduce significantly the relative voting power of each share of our common stock held by our current equityholders. Consequently, our equityholders as a group will have significantly less influence over the management and policies of the combined company after the merger than prior to the merger.
We have incurred and will continue to incur significant transaction costs in connection with the merger.
We have incurred and will continue to incur significant transaction costs in connection with the merger. We estimate that we will incur aggregate direct transaction costs of approximately $2.8 million associated with the merger and $0.5 million that we may pay on behalf of Achieve, as well as additional costs associated with the commencement of the combined company’s operation as a public company, which cannot be estimated accurately at this time.
The pendency of the merger could have an adverse effect on the trading price of our common stock and our business, financial condition, results of operations or business prospects.
While there have been no significant adverse effects to date, the pendency of the merger could disrupt our businesses in the following ways, including:
|
|
• |
the attention of our management may be directed toward completion of the merger and related matters and may be diverted from the day-to-day business operations, including identifying a collaboration partner to further the development of apatorsen and from other opportunities that otherwise might be beneficial to us; and |
|
|
• |
third parties may seek to terminate or renegotiate their relationships with us as a result of the merger, whether pursuant to the terms of their existing agreements with us or otherwise. |
Should they occur, any of these matters could adversely affect the trading price of our common stock or harm our financial condition, results of operations or business prospects.
As a result of the custirsen phase 3 trial results and the reductions in our workforce, we have only 12 employees remaining. If we are unable to retain the remaining employees, our ability to consummate the pending merger may be delayed or seriously jeopardized.
On February, October and November 2016, we announced workforce reductions, which have reduced the headcount to 12 remaining employees. Our cash conservation activities may yield unintended consequences, such as attrition beyond the planned reductions in workforce and reduced employee morale, which may cause the remaining 12 employees to seek alternate employment. Competition among biotechnology companies for qualified employees is intense, and the ability to retain the remaining employees is critical to our ability to effectively manage our resources and to consummate the pending merger. Additional attrition could have a material adverse effect on our business, including delaying the completion of wind down activities related to our custirsen clinical trials and related operations and increasing the time and funds required. In addition, as a result of the reduction in our workforce, we face an increased risk of employment litigation.
Risks Related to our Business
We have incurred losses since inception and anticipate that we will continue to incur losses for the foreseeable future. We have never had any products available for commercial sale and we may never achieve or sustain profitability.
We are a clinical-stage biopharmaceutical company, are not profitable, have incurred losses in each year since our inception and do not expect to become profitable in the foreseeable future. We have never had any products available for commercial sale, and we have not generated any revenue from product sales nor do we anticipate that we will generate revenue from product sales in the near future. Our revenue to date has been collaboration revenue under the Collaboration Agreement with Teva, which was terminated in April 2015. In addition, custirsen did not demonstrate its intended benefit in any phase 3 clinical trial and its development has been discontinued. Our other product candidate, apatorsen, is earlier in its development and will require a collaboration partner to fund the required additional development. We have not yet submitted any products for approval by regulatory authorities, and we continue to incur research and development and general and administrative expenses related to our operations. We expect to continue to incur losses for the foreseeable future. If we do not find a collaboration partner to fund additional development of apatorsen or apatorsen otherwise fails in clinical trials or does not gain regulatory approval, or if apatorsen does not achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
18
We cannot give any assurance that apatorsen will continue to be developed, receive regulatory approval or be successfully commercialized.
We conducted seven randomized phase 2 clinical trials evaluating apatorsen in several cancer indications. All but one of the phase 2 clinical trials for apatorsen failed to meet their pre-defined clinical endpoints. Completing additional clinical trials will be required to establish the safety and efficacy of this product candidate. We currently do not have sufficient capital to conduct additional clinical trials for apatorsen without a strategic partner, raising additional funds or completing a strategic transaction committed to the development of apatorsen. We are currently undertaking efforts to identify a third party to develop and, if approved, commercialize apatorsen. If we identify such a third party by August 2017 and our pending acquisition is completed, our stockholders will receive contingent value rights, or CVRs, to receive 80% of the consideration, less certain offsets, received by the combined company during the five-year period after the completion of the merger as a result of the achievement of certain clinical milestones, regulatory milestones, sales-based milestones and/or up-front payment milestones relating to apatorsen. We cannot give any assurance that we will be able to identify and enter into an agreement with a third party to develop and potentially commercialize apatorsen by August 2017, or if we do, that any consideration will ever be received by the combined company or distributed to our stockholders. If we are unable to enter into an agreement with a third party regarding the development of apatorsen by August 2017, the development of apatorsen may be delayed or terminated.
If we are able to enter into an agreement with a third party to develop apatorsen, the failure of apatorsen to be shown safe or effective in one or more indications could negatively impact the development of apatorsen in other indications, could result in the suspension or termination of apatorsen development and commercialization plans and could cause the CVRs to be of no or little value. Further, apatorsen consideration, if any, received beyond August 2022 would accrue to the benefit of the combined company stockholders generally and not to the CVR holders.
Our clinical development program for apatorsen may not receive regulatory approval either if apatorsen fails to demonstrate that it is safe and effective in clinical trials and consequently fail to obtain necessary approvals from the regulatory agencies, or if we have inadequate financial or other resources to advance apatorsen through the clinical trial process. If competitive products developed by third parties show significant benefit in the cancer indications in which we are developing apatorsen, any planned supportive or primary registration trials may be delayed, altered or not initiated and apatorsen may never receive regulatory approval. Any failure to obtain regulatory approval of apatorsen could have a material and adverse effect on our business.
Because we depend on financing from third parties for our operations, our business may fail if such financing becomes unavailable or is not available on commercially reasonable terms.
To date, we have financed our operations primarily through the sale of our equity securities and from payments we received pursuant to the Collaboration Agreement with Teva. In April 2015, our Collaboration Agreement with Teva was terminated, and we will not receive any future payments from Teva. We believe that our existing capital resources and interest on such resources will be sufficient to meet our current operating requirements for at least the next 12 months. However, if the timeline to complete the recently announced merger takes longer than anticipated or is not completed, we change our development plans or elect to further develop apatorsen, cannot find third-party collaborators to fund further development of apatorsen, our trials proceed slower or take longer than expected to complete, we acquire rights to new product candidates, do not successfully defend litigation or engage in commercialization and product launch activities, we will need additional capital sooner than we expect. Our future capital requirements will depend on many factors, including, without limitation:
|
|
• |
the timing of completion of the pending merger with Achieve; |
|
|
• |
whether we modify our development program for apatorsen, including terminating and starting new trials; |
|
|
• |
whether we are able to enter into additional third-party collaborative partnerships to develop and/or commercialize apatorsen on terms that are acceptable to us, or at all; |
|
|
• |
the scope and results of our clinical trials and preclinical studies; |
|
|
• |
our ability to forecast the cost of our ongoing development activities; |
|
|
• |
the availability of third parties to perform the key development tasks for apatorsen, including conducting preclinical studies and clinical trials and manufacturing apatorsen to be tested in those studies and trials and the associated costs of those services; |
19
|
|
• |
whether opportunities to acquire additional product candidates arise and the costs of acquiring and developing those product candidates; |
|
|
• |
the costs to defend, and the results of, litigation; and |
|
|
• |
whether we engage in commercialization and product launch activities. |
If we are unable to raise funds on acceptable terms when it becomes necessary to do so, we may not be able to continue developing apatorsen, acquire or develop additional product candidates or respond to competitive pressures or unanticipated requirements. For these reasons, any inability to raise additional funds when we require it could have a material adverse effect on our business.
We intend to partner with third-party collaborators with respect to the development and commercialization of apatorsen, and we cannot control whether we will be able to do so on favorable terms, if at all.
We are currently undertaking efforts to identify and enter into an agreement with a third party to fund and undertake the development and potential commercialization of apatorsen. If we are not able to do so by August 2017 and the pending merger is completed, the CVRs will be terminated and the CVR holders will not realize any value from the CVRs.
We will be competing with many other companies as we seek partners for apatorsen and may not be able to compete successfully against those companies. If we are not able to enter into collaboration arrangements for apatorsen, we would be required to undertake and fund further development, clinical trials, manufacturing and commercialization activities solely at our own expense and risk. If we are unable to finance and/or successfully execute those expensive activities, or we delay such activities due to capital availability, our business could be materially and adversely affected, and potential future product launch could be materially delayed, be less successful, or we may be forced to discontinue clinical development of our product candidate.
Clinical trials may not demonstrate a clinical benefit of apatorsen.
Positive results from preclinical studies and clinical trials, including any exploratory results from the apatorsen clinical trials conducted to date should not be relied on as evidence that on-going, amended, or later-stage or large-scale clinical trials will succeed.
We, or a collaboration partner, will be required to demonstrate with substantial evidence through well-controlled clinical trials that apatorsen is safe and effective for use in a diverse population before we or a collaboration partner can seek regulatory approvals for its commercial sale. Success in early clinical trials does not mean that future clinical trials will be successful because evaluation of apatorsen in later-stage clinical trials may fail to demonstrate sufficient safety and efficacy to the satisfaction of regulatory agencies, despite having progressed through initial clinical trials. For example, all our phase 3 clinical trials for custirsen failed to meet their clinical endpoints, even after encouraging results in earlier trials. Further, preliminary or top-line results from clinical trials may not be confirmed in final data, or may change materially.
Even after the completion of phase 3 clinical trials, regulatory agencies may disagree with our clinical trial design and our interpretation of data, and may require us to conduct additional clinical trials to demonstrate the efficacy of apatorsen.
We may choose to make amendments to ongoing studies for any reason including to analyze final top line data earlier than planned. Any future amendments may compromise the integrity of the clinical trial results and may not be acceptable to regulators.
We rely on third parties to manufacture and supply apatorsen and other agents used in our clinical trials and potential future commercial use. A decrease in the availability or quality of apatorsen or agents could increase clinical trial costs, delay or halt clinical development or regulatory approval or commercialization of apatorsen, resulting in additional losses and depriving us of potential product revenue.
We do not own or operate manufacturing facilities, and we depend on third-party contract manufacturers for production of apatorsen and rely on other companies and their manufacturers for other agents used in all of our clinical trials. We lack the resources and the capability to manufacture apatorsen ourselves. To date, our product candidates, including apatorsen have been manufactured in limited quantities for preclinical studies and clinical trials. All active pharmaceutical ingredients, or API, and drug product for our product candidates have been manufactured for us by third parties pursuant to a purchase order or short-term contract that has been fulfilled.
If, in the future, apatorsen is approved for commercial sale, we or any pharmaceutical partner that has licensed apatorsen, if any, may need to manufacture apatorsen in commercial quantities. We cannot provide assurance that the third-party manufacturers with which we have contracted in the past will have sufficient capacity to satisfy future manufacturing needs, that additional purchases of API or
20
drug product will be negotiated with these or alternative manufacturers on terms favorable to us, if at all, or that the pharmaceutical partner that has licensed apatorsen, if any, will have sufficient capacity or expertise to satisfy future needs.
Third-party manufacturers may fail to perform under their contractual obligations, or may fail to deliver the required commercial quantities of bulk API or finished drug product on a timely basis and at commercially reasonable prices. We have experienced manufacturing quality issues resulting in an unusable lot of one of our product candidates in the past. Any performance failure on the part of our contract manufacturers could delay clinical development or regulatory approval or commercialization of apatorsen, depriving us of potential product revenue and resulting in additional losses. If an alternate manufacturer is required to be identified and qualified, clinical trials, regulatory submissions, required approvals or commercialization of apatorsen may be delayed or suspended, which may cause higher costs and could prevent successful commercialization of apatorsen. If one or more replacement manufacturers capable of production at a reasonably favorable cost, in adequate volumes, of adequate quality and on a timely basis, cannot be identified, demand for apatorsen likely cannot be met and clinical trials could be delayed or we could lose potential revenue. The ability to replace an existing API manufacturer may be difficult because the number of potential manufacturers is limited to approximately five manufacturers, and regulatory agencies must inspect any replacement manufacturer and review information related to product produced at the manufacturer before they can begin manufacturing our product candidates. It may be difficult or impossible to identify and engage a replacement manufacturer on acceptable terms in a timely manner, if at all. We expect to continue to depend on third-party contract manufacturers for the foreseeable future.
Apatorsen requires precise, high-quality manufacturing. Any of our contract manufacturers will be subject to ongoing periodic unannounced inspection by regulatory agencies to ensure strict compliance with current Good Manufacturing Practices, or cGMP, and other applicable government regulations and corresponding standards. If a contract manufacturer fails to achieve and maintain high manufacturing standards in compliance with cGMP regulations, manufacturing errors may be experienced resulting in patient injury or death, product recalls or withdrawals, delays or interruptions of production or failures in product testing or delivery, delay or prevention of filing or approval of marketing applications for apatorsen, cost overruns or other problems that could seriously affect our business.
Significant manufacturing scale-up may require additional validation studies, which the regulatory agencies must review and approve. Additionally, any third-party manufacturers retained to manufacture apatorsen on a commercial scale must pass regulatory agencies’ pre-approval inspection for conformance to cGMP regulations before approval of apatorsen can be obtained. If manufacturing capacity for apatorsen in conformance with cGMP regulations is not successfully increased, the regulatory approval or commercial launch of apatorsen may be delayed or there may be a shortage in supply.
We also rely on third parties for the provision of other agents used in our clinical trials, and in some circumstances these agents are provided to us at no cost. We have no assurance that these third-parties will continue to provide their products to us at no cost.
If our competitors develop and market products that are more effective, safer or less expensive than apatorsen, our clinical trials and commercial opportunities will be negatively affected.
The life sciences industry is highly competitive, and we face significant competition from many pharmaceutical, biopharmaceutical and biotechnology companies that are researching and marketing products designed to address cancer indications for which apatorsen is currently being developed or for which apatorsen may be developed in the future. We are aware of several other companies that are developing therapeutics that seek to promote tumor cell death. Several therapies have been recently approved by the FDA in indications for which apatorsen may be developed in the future, and we expect more to be approved in the future.
Substantial advancements in the treatment of cancer have occurred in the past two years and new products from our competitors have been approved for marketing on the basis of showing a survival advantage. Apatorsen may be developed in the future by a collaboration partner in any number of cancer indications, including in bladder cancer. Any product we may develop in the future is likely to face competition from other drugs and therapies. Many of our competitors have significantly greater financial, manufacturing, marketing and drug development resources than we do. Large pharmaceutical companies, in particular, have extensive experience in clinical testing and in obtaining regulatory approvals for drugs. These companies also have significantly greater research and marketing capabilities than we do. In addition, many universities and private and public research institutes are, or may become, active in cancer research, and develop products that may directly compete with ours. If our competitors market products that are more effective, safer or less expensive than our future product candidates, if any, or that reach the market sooner than our future product candidates, if any, we may not achieve commercial success.
If new therapies become broadly used, additional clinical trials of apatorsen in combination with these new therapies may be required to demonstrate safety and efficacy of the combination. Additional trials will delay the development of apatorsen and
21
increase our costs. The failure of apatorsen to work in combination with these new therapies would have an adverse effect on our business.
As new therapies are developed, these therapies will need to be assessed to determine whether to conduct clinical trials of apatorsen in combination with them to demonstrate safety and efficacy of the combination. If it is determined appropriate to conduct additional clinical trials of apatorsen in combination with these new therapies, the development of apatorsen will be delayed and our costs will be increased. If these clinical trials generate safety concerns or lack of efficacy, our business would be adversely affected.
We rely, in part, on third parties to conduct our clinical trial for apatorsen and may rely on third parties to conduct future clinical trials, if any. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, regulatory approval for or commercialization of apatorsen may not be obtained.
To implement our product development strategies, we rely on third parties, such as collaborators, contract research organizations, medical institutions, clinical investigators and contract laboratories, to conduct clinical trials of apatorsen. Although we rely on third parties to conduct our clinical trials, we are responsible for ensuring that each of our clinical trials is conducted in accordance with our development plan and protocol. Moreover, regulatory agencies require us to comply with regulations and standards, commonly referred to as Good Clinical Practices, or GCPs, for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate and that the clinical trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on third parties does not relieve us of these responsibilities and requirements. If the third parties conducting our clinical trials do not perform their contractual duties or obligations, do not meet expected deadlines or need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to GCPs or for any other reason, we may need to enter into new arrangements with alternative third parties and our clinical trials may be extended, delayed or terminated. In addition, a failure by such third parties to perform their obligations in compliance with GCPs may cause our clinical trials to fail to meet regulatory requirements, which may require us to repeat our clinical trials.
Our clinical trial may be suspended or terminated at any time, including by regulatory agencies, a Data Safety Monitoring Board overseeing the clinical trial at issue, by a clinical trial site or investigator, or by us. Any failure or significant delay in completing our clinical trial for apatorsen could materially harm the commercial prospects for apatorsen.
We do not know whether our clinical trial for apatorsen will proceed or be completed on schedule, if at all, or whether we will be able to identify a collaboration partner to fund and manage any future preclinical studies or clinical trials, as applicable. The completion of our clinical trial currently in progress could also be substantially delayed or prevented by several factors, including:
|
|
• |
delay or failure to complete the merger with Achieve ; |
|
|
• |
the strategic development plan of the combined company following completion of the pending merger; |
|
|
• |
termination of the clinical trial by us, by one or more clinical trial sites, investigators, data safety monitoring boards, granting or regulatory agencies; |
|
|
• |
delay or failure to obtain sufficient manufacturing supply of apatorsen, or expiration of our existing supply of apatorsen prior to completing our ongoing clinical trial; |
|
|
• |
lack of efficacy evidenced during the clinical trial; |
|
|
• |
slower than expected final analysis of the clinical trial data; |
|
|
• |
failure of patients to complete the clinical trial; |
|
|
• |
unforeseen safety issues; |
|
|
• |
inability or unwillingness of patients or medical investigators to follow the clinical trial protocol; |
|
|
• |
inability to monitor patients adequately during or after treatment; |
|
|
• |
introduction of competitive products that may impede our ability to retain patients in the clinical trial; and |
|
|
• |
delay in submission or acceptance of protocol amendments, if any. |
Apatorsen may cause undesirable and potentially serious side effects during clinical trials that could delay or prevent its regulatory approval or commercialization.
Since patients in our clinical trials have advanced stages of cancer, we expect that additional adverse events, including serious adverse events, will occur.
22
Undesirable side effects caused by apatorsen could cause us or regulatory authorities to amend, interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by regulatory agencies for any or all targeted indications or decrease the competitive opportunity of apatorsen which may decrease sales potential. This, in turn, could prevent commercialization of apatorsen and generating revenue from its sale. In addition, if apatorsen receives marketing approval and we or others later identify undesirable side effects caused by the product:
|
|
• |
regulatory authorities may withdraw their approval of the apatorsen; |
|
|
• |
apatorsen may be recalled, or a change in the way it is administered may be required, additional clinical trials may be required or a change in the labeling of apatorsen may be necessary; |
|
|
• |
apatorsen may become less competitive and sales may decrease; and |
|
|
• |
our reputation may suffer. |
Any one or a combination of these events could prevent achievement or maintenance of market acceptance of apatorsen or could substantially increase the costs and expenses of commercializing the product, which in turn could delay or prevent the generation of significant revenue from the sale of the product. Historic events have raised questions about the safety of other companies’ marketed drugs and may result in increased cautiousness by regulatory agencies in reviewing new drugs based on safety, efficacy or other regulatory considerations and may result in significant delays in obtaining regulatory approvals, additional clinical trials being required, or more stringent product labeling requirements. Any delay in obtaining, or the inability to obtain, applicable regulatory approvals would prevent commercialization of apatorsen.
If we were to be successfully sued related to our products or operations, we could face substantial liabilities that may exceed our resources.
We may be held liable if any of our products or operations cause injury or death or are found otherwise unsuitable during product testing, manufacturing, marketing or sale. These risks are inherent in the development of pharmaceutical products. We currently maintain commercial general and umbrella liability policies with combined limits of $10.0 million per occurrence and in the aggregate, in addition to a $10.0 million per claim and annual aggregate product liability insurance policy related to our clinical trials consistent with industry standards. When necessary for our products, we intend to obtain additional product liability insurance. Insurance coverage may be prohibitively expensive, may not fully cover potential liabilities or may not be available in the future. Inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the commercialization of our products. If we were to be sued for any injury caused by or associated with our products or operations, the litigation could consume substantial time and attention of our management, and the resulting liability could exceed our total assets.
Even if regulatory approval to market apatorsen is received, the market may not be receptive to the product.
Even if apatorsen obtains regulatory approval, it may not gain market acceptance among physicians, patients, healthcare payors and/or the medical community. We believe that the degree of market acceptance will depend on a number of factors, including:
|
|
• |
efficacy, safety and tolerability of apatorsen; |
|
|
• |
timing of market introduction of competitive products; |
|
|
• |
availability of coverage and reimbursement from government and other third-party payors; |
|
|
• |
potential advantages or disadvantages over alternative treatments; |
|
|
• |
strength of marketing and distribution support; |
|
|
• |
price of our apatorsen, both in absolute terms and relative to alternative treatments; and |
|
|
• |
sequencing of available products. |
If our future product candidates fail to achieve market acceptance, we may not be able to generate significant revenue or achieve or sustain profitability.
The successful commercialization of apatorsen will depend in part on the extent to which governmental authorities and health insurers establish adequate coverage and reimbursement levels and pricing policies.
23
Successful sales of apatorsen will depend, in part, on the extent to which coverage and reimbursement for the product will be available from government and health administration authorities, private health insurers and other third-party payors. To manage healthcare costs, many governments and third-party payors increasingly scrutinize the pricing of new products and require greater levels of evidence of favorable clinical outcomes and cost-effectiveness before extending coverage. In light of such challenges to prices, we cannot be sure that coverage for apatorsen will be obtained or, if available, that the reimbursement rates will be adequate. If adequate levels of coverage and reimbursement for apatorsen cannot be attained, its marketability will be negatively and materially impacted.
Moreover, eligibility for coverage and reimbursement does not imply that a drug will be paid for in all cases or at a rate that covers costs, including research, development, manufacture, sale and distribution. In addition, obtaining and maintaining adequate coverage and reimbursement status is time-consuming and costly. Third party payors may deny coverage and reimbursement status altogether of a given drug product, or cover the product but may also establish prices at levels that are too low to enable us to realize an appropriate return on our investment in product development. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage for the product. Because the rules and regulations regarding coverage and reimbursement change frequently, in some cases at short notice, even when there is favorable coverage and reimbursement, future changes may occur that adversely impact the favorable status.
The unavailability or inadequacy of third-party coverage and reimbursement could have a material adverse effect on the market acceptance of any of our future products and the future revenues we may expect to receive from those products. In addition, we are unable to predict what additional legislation or regulation relating to the healthcare industry or third-party coverage and reimbursement may be enacted in the future, or what effect such legislation or regulation would have on our business.
We may fail to acquire and develop additional products or product candidates at all or on commercially reasonable terms.
We currently do not have internal discovery capabilities and depend on pharmaceutical and biotechnology companies and other researchers to sell or license products or product candidates to us. If we are unable to complete the merger with Achieve, we may be required to identify alternative sources of product candidates.
To successfully build a product pipeline, we would be required to identify, select and acquire pharmaceutical product candidates. Proposing, negotiating and implementing an economically viable product acquisition or license is a lengthy and complex process. We compete for partnering arrangements and license agreements with pharmaceutical and biotechnology companies and academic research institutions. Our competitors may have stronger relationships with third parties with whom we are interested in collaborating and/or may have more established histories of developing and commercializing products. As a result, our competitors may have a competitive advantage in entering into partnering arrangements with such third parties. In addition, even if we find promising product candidates, and generate interest in a partnering or strategic arrangement to acquire such product candidates, we may not be able to acquire rights to additional product candidates or approved products on terms that we find acceptable, if at all. If we fail to acquire and develop product candidates from others, we may be unable to grow our business.
We expect that any product candidate that we acquire rights to will require additional development efforts prior to commercial sale, including extensive clinical evaluation and approval by regulatory agencies. All product candidates are subject to the risks of failure inherent in pharmaceutical product development, including the possibility that the product candidate will not be shown to be sufficiently safe and effective for approval by regulatory authorities. Even if the product candidates are approved, we can make no assurance that we would be capable of economically producing the product or that the product would be commercially successful.
We may be adversely affected if our controls over financial reporting fail or are circumvented.
We regularly review and update our internal controls, disclosure controls and procedures, and corporate governance policies. In addition, although not required, we have chosen under the Sarbanes Oxley Act of 2002 to report annually on our internal control over financial reporting. If it were to be determined that our internal control over financial reporting is not effective, such shortcoming could have an adverse effect on our business and financial results and the price of our common stock could be negatively affected. This reporting requirement could also make it more difficult or more costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. Any system of internal controls, however well designed and operated, is based in part on certain assumptions and can provide only reasonable, not absolute, assurances that the objectives of the system are met. Any failure or circumvention of the controls and procedures or failure to comply with regulation concerning control and procedures could have a material effect on our business, results of operation and financial condition. Any of these events could result in an adverse reaction in the financial marketplace due to a loss of investor confidence in the reliability of our financial statements, which ultimately could negatively affect the market price of our shares, increase the volatility of our stock price and adversely affect our ability to raise additional funding. The effect of these events could also make it more difficult for us to attract and retain qualified persons to serve on our Board and our Board committees and as executive officers.
24
Risks Related to Our Intellectual Property
Our proprietary rights may not adequately protect apatorsen.
Our commercial success will depend in part on our ability to obtain patents and/or regulatory exclusivity and maintain adequate protection for apatorsen in the United States and other countries. We or a collaboration partner, if any, will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that apatorsen is covered by valid and enforceable patents or are effectively maintained as trade secrets.
We and/or a collaboration partner, if any, may apply for additional patents covering apatorsen as we deem appropriate. We or our collaboration partner, if any, may, however, fail to apply for patents on important technologies or apatorsen in a timely fashion, if at all. Our existing patents and any future patents we or our collaboration partner, if any, obtain may not be sufficiently broad to prevent others from practicing our technologies or from developing competing products and technologies. In addition, we do not always control the patent prosecution of subject matter that we license from others. Accordingly, we are sometimes unable to exercise a significant degree of control over such intellectual property as we would over our own.
Moreover, the patent positions of biopharmaceutical companies are highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. As a result, the validity and enforceability of our patents cannot be predicted with certainty. In addition, the U.S. Supreme Court has revised certain tests regarding granting patents and assessing the validity of patents to make it more difficult to obtain patents. As a consequence, issued patents may be found to contain invalid claims according to the revised standards. Some of our patents or those of collaborators may be subject to challenge and subsequent invalidation or significant narrowing of claim scope in a re-examination proceeding, or during litigation, under the revised criteria. We cannot guarantee that:
|
|
• |
we or our licensors were the first to make the inventions covered by each of our issued patents and pending patent applications; |
|
|
• |
we or our licensors were the first to file patent applications for these inventions; |
|
|
• |
others will not independently develop similar or alternative technologies or duplicate any of our technologies; |
|
|
• |
any of our or our licensors’ pending patent applications will result in issued patents; |
|
|
• |
any of our or our licensors’ patents will be valid or enforceable; |
|
|
• |
any patents issued to us or our licensors and collaboration partners will provide us with any competitive advantages, or will not be challenged by third parties; and |
|
|
• |
we will develop additional proprietary technologies that are patentable, or the patents of others will not have an adverse effect on our business. |
The actual protection afforded by a patent varies on a product-by-product basis, from country to country and depends on many factors, including the type of patent, the scope of its coverage, the availability of regulatory related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patents. Our ability or the ability of a collaboration partner, if any, to maintain and solidify our proprietary position for apatorsen will depend on our success in obtaining effective claims and enforcing those claims once granted. Our issued patents and those that may issue in the future, or those licensed to us or collaboration partners, if any, may be challenged, invalidated, unenforceable or circumvented, and the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar products. Due to the extensive amount of time required for the development, testing and regulatory review of a potential product, it is possible that, before apatorsen can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
We also rely on trade secrets to protect some of our technology, especially where it is believed that patent protection is not appropriate or obtainable. However, trade secrets are difficult to maintain. While we use reasonable efforts to protect our trade secrets, our or our collaboration partners’ employees, consultants, contractors or scientific and other advisors may unintentionally or willfully disclose our proprietary information to competitors. Enforcement of claims that a third party has illegally obtained and is using trade secrets is expensive, time consuming and uncertain. In addition, non-U.S. courts are sometimes less willing than U.S. courts to protect trade secrets. If our competitors independently develop equivalent knowledge, methods and know-how, we would not be able to assert our trade secrets against them and our business could be harmed.
We may not be able to protect our intellectual property rights throughout the world.
25
Filing, prosecuting and defending patents on apatorsen, when and if we have any, in every jurisdiction would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we or our licensors have not obtained patent protection to develop their own products. These products may compete with our products, when and if we have any, and may not be covered by any of our or our licensors’ patent claims or other intellectual property rights.
The laws of some non-U.S. countries do not protect intellectual property rights to the same extent as the laws of the United States, and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biotechnology and/or pharmaceuticals, which could make it difficult for us to stop the infringement of our patents. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
We may become involved in disputes with past or potential future collaborators over intellectual property ownership, and publications by our research collaborators and scientific advisors could impair our ability to obtain patent protection or protect our proprietary information, which, in either case, could have a significant effect on our business.
Inventions discovered under research, material transfer or other such collaborative agreements may become jointly owned by us and the other party to such agreements in some cases and the exclusive property of either party in other cases. Under some circumstances, it may be difficult to determine who owns a particular invention, or whether it is jointly owned, and disputes could arise regarding ownership of those inventions. These disputes could be costly and time consuming and an unfavorable outcome could have a significant adverse effect on our business if we were not able to protect or license rights to these inventions. In addition, our research collaborators and scientific advisors generally have contractual rights to publish our data and other proprietary information, subject to our prior review. Publications by our research collaborators and scientific advisors containing such information, either with our permission or in contravention of the terms of their agreements with us, may impair our ability to obtain patent protection or protect our proprietary information, which could significantly harm our business.
The intellectual property protection for apatorsen depends on third parties.
We have exclusively licensed from UBC certain issued patents and pending patent applications covering the respective antisense sequences underlying apatorsen and its commercialization and use, and we have licensed from Ionis certain issued patents and pending patent applications directed to product compositions and chemical modifications used in apatorsen for commercialization, use and the manufacturing thereof. We have also received a sublicense from Ionis under certain third-party patent portfolios directed to such modifications.
The patents and pending patent applications underlying our licenses do not fully cover all potential modifications and uses of apatorsen. In the case of patents and patent applications licensed from Ionis, we do not have and have not had any control over the filing, prosecution or enforcement of these patents or patent applications. We cannot be certain that such prosecution efforts have been or will be conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents. We also cannot be assured that our licensors or their respective licensing partners will agree to enforce any such patent rights at our request or devote sufficient efforts to attain a desirable result. Any failure by our licensors or any of their respective licensing partners to properly protect the intellectual property rights relating to apatorsen could have a material adverse effect on our financial condition and results of operation.
If we breach any of the agreements under which we license rights to apatorsen or technology from third parties, we could lose license rights that are important to our business. Certain of our license agreements may not provide an adequate remedy for a breach by the licensor.
We license the development and commercialization rights for apatorsen. Under such licenses, we are subject to various obligations such as sublicensing, royalty and milestone payments, annual maintenance fees, limits on sublicensing, insurance obligations and the obligation to use commercially reasonable best efforts to develop and exploit the licensed technology. If we fail to comply with any of these obligations or otherwise breach these agreements, our licensors may have the right to terminate the license in whole or in part or to terminate the exclusive nature of the license. We may also become involved in disputes with current or former licensors regarding the meaning of certain terms in the license agreements, including terms related to royalty and milestone payments and termination, which may result in costly and time consuming litigation. Loss of any of these licenses or the exclusivity rights provided by the licenses, or disputes with current or former licensors, could harm our financial condition and results of operations. In addition, certain of our license agreements with UBC eliminate our ability to obtain money damages in respect of certain claims against UBC.
26
The patent protection for apatorsen may expire before we are able to maximize its commercial value, which may subject us to increased competition and reduce or eliminate our opportunity to generate product revenue.
The patents for apatorsen have varying expiration dates and, when these patents expire, we may be subject to increased competition and we may not be able to recover our development costs. For example, certain of the U.S. patents directed to apatorsen and its use that have been licensed from UBC are expected to expire in 2023. In some of the larger economic territories, such as the United States and Europe, patent term extension/restoration may be available to compensate for time taken during aspects of the product candidate’s regulatory review. We cannot, however, be certain that an extension will be granted or, if granted, what the applicable time period or the scope of patent protection afforded during any extended period will be. In addition, even though some regulatory agencies may provide some other exclusivity for a product candidate under its own laws and regulations, we may not be able to qualify the product candidate or obtain the exclusive time period.
If we are unable to obtain patent term extension/restoration or some other exclusivity, we could be subject to increased competition and our opportunity to establish or maintain product revenue could be substantially reduced or eliminated. Furthermore, we may not have sufficient time to recover our development costs prior to the expiration of our U.S. and non-U.S. patents.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and we may be unable to protect our rights to, or use of, our technology.
If we choose to go to court to stop someone else from using the inventions claimed in our patents or our licensed patents, that individual or company has the right to ask the court to rule that these patents are invalid and/or should not be enforced against that third party. These lawsuits are expensive and would consume time and other resources even if we were successful in stopping the infringement of these patents. In addition, there is a risk that the court will decide that these patents are invalid or unenforceable and that we do not have the right to stop the other party from using the inventions. The U.S. Supreme Court has revised certain tests regarding granting patents and assessing the validity of patents to make it more difficult to obtain patents. Some of our issued patents may be subject to challenge and subsequent invalidation under the revised criteria. There is also the risk that, even if the validity or unenforceability of these patents is upheld, the court will narrow the scope of our claim or will refuse to stop the other party on the grounds that such other party’s activities do not infringe our rights.
If we wish to use the technology or compound claimed in issued and unexpired patents owned by others, we will need to obtain a license from the owner, enter into litigation to challenge the validity or enforceability of the patents or incur the risk of litigation in the event that the owner asserts that we infringed its patents. The failure to obtain a license to technology or the failure to challenge an issued patent that we may require to discover, develop or commercialize apatorsen may have a material adverse effect on us.
If a third party asserts that we infringed its patents or other proprietary rights, we could face a number of risks that could seriously harm our results of operations, financial condition and competitive position, including:
|
|
• |
patent infringement and other intellectual property claims, which would be costly and time consuming to defend, whether or not the claims have merit, and which could delay the regulatory approval process and divert management’s attention from our business; |
|
|
• |
substantial damages for past infringement, which we may have to pay if a court determines that apatorsen or our technologies infringe a competitor’s patent or other proprietary rights; |
|
|
• |
a court prohibiting us from selling or licensing our technologies or future drugs unless the third party licenses its patents or other proprietary rights to us on commercially reasonable terms, which it is not required to do; and |
|
|
• |
if a license is available from a third party, we may have to pay substantial royalties or lump-sum payments or grant cross licenses to our patents or other proprietary rights to obtain that license. |
The biotechnology industry has produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that apatorsen or methods of use either do not infringe the patent claims of the relevant patent, and/or that the patent claims are invalid, and/or that the patent is unenforceable and we may not be able to do this. Proving invalidity, in particular, is difficult since it requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
U.S. patent laws as well as the laws of some foreign jurisdictions provide for provisional rights in published patent applications beginning on the date of publication, including the right to obtain reasonable royalties, if a patent subsequently issues and certain other conditions are met.
27
Because some patent applications in the United States may be maintained in secrecy until the patents are issued, because patent applications in the United States and many foreign jurisdictions are typically not published until 18 months after filing and because publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our licensors’ issued patents or our pending applications or our licensors’ pending applications, or that we or our licensors were the first to invent the technology.
Patent applications filed by third parties that cover technology similar to ours may have priority over our or our licensors’ patent applications and could further require us to obtain rights to issued patents covering such technologies. If another party files a U.S. patent application on an invention similar to ours, we may elect to participate in or be drawn into an interference proceeding declared by the U.S. Patent and Trademark Office to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful, resulting in a loss of our U.S. patent position with respect to such inventions. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations. We cannot predict whether third parties will assert these claims against us or against the licensors of technology licensed to us, or whether those claims will harm our business. If we are forced to defend against these claims, whether they are with or without any merit and whether they are resolved in favor of or against us or our licensors, we may face costly litigation and diversion of management’s attention and resources. As a result of these disputes, we may have to develop costly non-infringing technology, or enter into licensing agreements. These agreements, if necessary, may be unavailable on terms acceptable to us, if at all, which could seriously harm our business or financial condition.
We may be subject to damages resulting from claims that we, or our employees or consultants, have wrongfully used or disclosed alleged trade secrets of third parties.
Many of our employees were previously employed, and certain of our consultants are currently employed, at universities or biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we have not received any claim to date, we may be subject to claims that these employees or consultants or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of these current or former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. We may be subject to claims that employees of our partners or licensors of technology licensed by us have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. We may become involved in litigation to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel.
Risks Related to our Common Stock
The price for our common stock is volatile.
The market prices for our common stock and that of emerging life science companies generally have historically been highly volatile. For example, after the announcement of data from recent custirsen and apatorsen clinical trials, we experienced significant decreases in our stock price. Future announcements concerning us, our pending merger, the results of our clinical trials or our competitors may also have a significant effect on the market price of our common stock. The stock markets also experience significant price and volume fluctuation unrelated to the operating performance of particular companies. These market fluctuations may also adversely affect the market price of our common stock.
An increase in the market price of our common stock, which is uncertain and unpredictable, may be the sole source of gain from an investment in our common stock. An investment in our common stock may not be appropriate for investors who require dividend income. We have never declared or paid cash dividends on our capital stock and do not anticipate paying any cash dividends on our capital stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and growth of our business. As a result, capital appreciation, if any, of our common stock will be the sole source of gain for stockholders for the foreseeable future. Accordingly, an investment in our common stock may not be appropriate for investors who require dividend income or investors who are not prepared to bear a significant risk of losses from such an investment.
The price of our common stock does not meet the requirements for continued listing on The NASDAQ Capital Market. If we fail to regain compliance with the minimum listing requirements, our common stock will be subject to delisting. Our ability to complete the pending merger or publicly or privately sell equity securities and the liquidity of our common stock could be adversely affected if our common stock is delisted.
28
The continued listing standards of The NASDAQ Capital Market require, among other things, that the minimum bid price of a listed company’s stock be at or above $1.00. If the minimum bid price is below $1.00 for a period of more than 30 consecutive trading days, the listed company will fail to be in compliance with The NASDAQ Capital Market’s listing rules and, if it does not regain compliance within the grace period, will be subject to delisting. As previously reported, on August 22, 2016, we received a notice from the NASDAQ Listing Qualifications Department notifying us that for 30 consecutive trading days, the bid price of our common stock had closed below the minimum $1.00 per share requirement. In accordance with The NASDAQ Capital Market’s listing rules, we were afforded 180 calendar days, or until February 21, 2017, to regain compliance with the bid price requirement. In order to regain compliance, the bid price of our common stock must close at a price of at least $1.00 per share for a minimum of 10 consecutive trading days. On February 22, 2017, we received a second notice from the NASDAQ Listing Qualifications Department notifying us that we had not regained compliance with the bid price requirement during the 180 calendar days, and that we may be eligible for an additional 180-day compliance period if we meet the market value of publicly held shares requirement for continued listing, all other initial inclusion requirements for The NASDAQ Capital Market, except for the bid price requirement, and provide written notice that we intend to regain compliance with the bid price requirement during the second 180-day compliance period, by effecting a reverse stock split if necessary. We believe we are eligible for the additional 180-day compliance period, and intend to meet the bid price requirement by effecting a reverse stock split upon the completion of our pending merger.
If we fail to regain compliance, our common stock will be subject to delisting. Delisting from The NASDAQ Capital Market could adversely affect our ability to raise additional financing through the public or private sale of equity securities, would significantly affect the ability of investors to trade our securities and would negatively affect the value and liquidity of our common stock. Delisting could also have other negative results, including the potential loss of confidence by employees, the loss of institutional investor interest and fewer business development opportunities. Delisting would also prevent us from satisfying a closing condition for the pending merger, and, in such event, Achieve may elect not to consummate the merger. In addition, the combined company must submit a new application for listing on The NASDAQ Capital Market after the merger pursuant to the reverse merger rules, and the combined company will need to meet NASDAQ’s minimum listing requirements.
We are at risk of securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities, including in circumstances where such declines occur in close proximity to the announcement of clinical trial results, as well as following certain significant business transactions, such as the announcement of a merger. This risk is especially relevant for us because we recently announced a pending merger with Achieve. Additionally, our stock price and those of other biotechnology and biopharmaceutical companies have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business. Any stockholder litigation challenging the pending merger may also delay completion of the merger in the expected timeframe or altogether.
If we raise additional capital, the terms of the financing transactions may cause dilution to existing stockholders or contain terms that are not favorable to us.
To date, our sources of cash have been limited primarily to proceeds from the private or public placement of our securities and reimbursement for custirsen-related development expenses from our prior strategic collaboration with Teva, which terminated in April 2015. In the future, we may seek to raise additional financing through private placements or public offerings of our equity or debt securities. We cannot be certain that additional funding will be available on acceptable terms, if at all. To the extent that we raise additional financing by issuing equity securities, we may do so at a price per share that represents a discount to the then-current per share trading price of our common stock and our stockholders may experience significant dilution. Any debt financing, if available, may involve restrictive covenants, such as limitations on our ability to incur additional indebtedness, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely affect our ability to conduct our business.
Risks Related to Our Industry
There is a high risk that our drug development activities will not result in commercial products.
We or a collaborator, if any, will need to complete significant additional clinical trials before we or they can demonstrate that apatorsen is safe and effective to the satisfaction of regulatory agencies. Clinical trials are expensive and uncertain processes that take years to complete. Failure can occur at any stage of the process, and successful early clinical trials do not ensure that later clinical trials will be successful. In later-stage clinical trials, apatorsen may fail to show desired efficacy and safety traits despite having progressed through initial clinical trials. For example, all of our phase 3 clinical trials for custirsen and all but one of our phase 2 trials for apatorsen failed to meet their clinical endpoints, even after positive results in earlier trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier clinical trials. In addition, a clinical trial may prove successful with respect to a secondary objective, but fail to demonstrate clinically
29
significant benefits with respect to a primary objective. Failure to satisfy a primary objective in a phase 3 clinical trial (registration trial) would generally mean that a product candidate would not receive regulatory approval.
The regulatory approval process is expensive, time consuming and uncertain and may prevent us from obtaining approvals for the commercialization of some or all of our product candidates.
The research, testing, manufacturing, labeling, approval, selling, marketing and distribution of drug products are subject to extensive regulation by regulatory agencies, which regulations differ from country to country. We are not permitted to market our product candidate in the United States until we receive approval of an application for market approval from regulatory agencies. We have not submitted an application for or received marketing approval for our apatorsen. Obtaining approval of an application for market approval can be a lengthy, expensive and uncertain process. In addition, failure to comply with regulatory agencies’ requirements may, either before or after product approval, if any, subject us to administrative or judicially imposed sanctions, including:
|
|
• |
restrictions on the products, manufacturers or manufacturing process; |
|
|
• |
warning letters; |
|
|
• |
civil and criminal penalties; |
|
|
• |
injunctions; |
|
|
• |
suspension or withdrawal of regulatory approvals; |
|
|
• |
product seizures, detentions or import bans; |
|
|
• |
voluntary or mandatory product recalls and publicity requirements; |
|
|
• |
total or partial suspension of production; |
|
|
• |
imposition of restrictions on operations, including costly new manufacturing requirements; and |
|
|
• |
refusal to approve pending applications for market approval or supplements to approved applications for market approval. |
Regulatory approval of an application for market approval or application for market approval supplement is not guaranteed, and the approval process is expensive and may take several years. Regulatory agencies also have substantial discretion in the drug approval process. Despite the time and expense exerted, failure can occur at any stage, and we or a collaborator, if any, could encounter problems that could cause us or them to abandon clinical trials or to repeat or perform additional preclinical studies and clinical trials. The number of preclinical studies and clinical trials that will be required for regulatory agencies’ approval varies depending on the drug candidate, the disease or condition that the drug candidate is designed to address, and the regulations applicable to any particular drug candidate. Regulatory agencies can delay, limit or deny approval of a drug candidate for many reasons, including:
|
|
• |
a drug candidate may not be deemed safe or effective; |
|
|
• |
regulatory agencies may not find the data from preclinical studies and/or clinical trials sufficient; |
|
|
• |
regulatory agencies might not approve our third-party manufacturer’s processes or facilities; |
|
|
• |
regulatory agencies may change its approval policies or adopt new regulations; and |
|
|
• |
third-party products may enter the market and change approval requirements. |
Even if we or a collaborator, if any, obtains regulatory approvals for apatorsen, the terms of approvals and ongoing regulation of apatorsen may limit how we or a collaborator, if any, manufactures and markets apatorsen, which could materially affect our ability to generate revenue.
If apatorsen was approved, it and its manufacturer will be subject to continual review. Any regulatory approval that we or a collaborator, if any, receives for apatorsen is likely to be subject to limitations on the indicated uses for which the end product may be marketed, or include requirements for potentially costly post-approval follow-up clinical trials. In addition, if regulatory agencies approve apatorsen, the labeling, packaging, adverse event reporting, storage, advertising and promotion for the end product will be subject to extensive regulatory requirements. The manufacturers of apatorsen, when and if it has any, will also be required to comply with cGMP regulations, which include requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation. Further, regulatory agencies must approve these manufacturing facilities before they can be used to manufacture our product, when and if apatorsen has any, and these facilities are subject to ongoing regulatory inspection. If the manufacturer fails to comply with the regulatory requirements of regulatory agencies, or if previously unknown problems with apatorsen are discovered, we could be subject to administrative or judicially imposed sanctions, including:
|
|
• |
restrictions on the product, manufacturers or manufacturing process; |
30
|
|
• |
civil or criminal penalties or fines; |
|
|
• |
injunctions; |
|
|
• |
product seizures, detentions or import bans; |
|
|
• |
voluntary or mandatory product recalls and publicity requirements; |
|
|
• |
|
|
• |
total or partial suspension of production; |
|
|
• |
imposition of restrictions on operations, including costly new manufacturing requirements; and |
|
|
• |
refusal to approve pending applications for market approval or supplements to approved applications for market approval. |
In addition, regulatory agencies may change their policies and additional regulations may be enacted that could prevent or delay regulatory approval of apatorsen. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States, Canada or abroad. If we are not able to maintain regulatory compliance, we would likely not be permitted to market apatorsen and we may not achieve or sustain profitability.
If government and third-party payors fail to provide coverage and adequate reimbursement rates for apatorsen, our revenue and potential for profitability will be reduced.
In the United States and elsewhere, our product revenue will depend principally on the reimbursement rates established by third-party payors, including government health administration authorities, managed-care providers, public health insurers, private health insurers and other organizations. These third-party payors are increasingly challenging the price, and examining the cost-effectiveness, of medical products and services. In addition, significant uncertainty exists as to the reimbursement status, if any, of newly approved drugs, pharmaceutical products or product indications. We or a collaborator, if any, may need to conduct post-marketing clinical trials in order to demonstrate the cost-effectiveness of apatorsen, if any. Such clinical trials may require us or a collaborator, if any, to commit a significant amount of management time and financial and other resources. If reimbursement of such product is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels, our revenue could be reduced.
In some countries other than the United States, particularly the countries of the European Union and Canada, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, obtaining pricing approval from governmental authorities can take six to 12 months or longer after the receipt of regulatory marketing approval of a product for an indication. To obtain reimbursement or pricing approval in some countries, we or a collaborator, if any, may be required to conduct a clinical trial that compares the cost-effectiveness of apatorsen to other available therapies. If reimbursement of such product candidate is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels, our revenue could be reduced.
Domestic and foreign governments continue to propose and pass legislation designed to reduce the cost of healthcare, including drugs. In the United States, there have been, and we expect that there will continue to be, federal and state proposals to implement similar governmental control. In addition, increasing emphasis on managed care in the United States will continue to put pressure on the pricing of pharmaceutical products. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, PPACA, became law in the United States. PPACA substantially changes the way healthcare is financed by both governmental and private insurers and significantly affects the pharmaceutical industry.
We anticipate that the PPACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and downward pressure on the price for any approved product, and could seriously harm our prospects. In addition, the Medicare and Medicaid program and state healthcare laws and regulations may also be modified to change the scope of covered products and/or reimbursement methodology. Cost control initiatives could decrease the established reimbursement rates that we receive for apatorsen in the future, which would limit our revenue and profitability. Legislation and regulations affecting the pricing of pharmaceutical products, including apatorsen, may change at any time, which could further limit or eliminate reimbursement rates for apatorsen or other product candidates.
Failure to obtain regulatory approval outside of the United States and Canada would prevent us from marketing our product candidates abroad.
We or a collaborator may market apatorsen outside of the United States and Canada. In order to market apatorsen in the European Union and many other non-North American markets, we or a collaborator, if any, must obtain separate regulatory approvals. We have
31
had limited interactions with non-North American regulatory authorities. Approval procedures vary among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. Approval by the FDA or other regulatory authorities does not ensure approval by regulatory authorities in other countries, and approval by one or more non-North American regulatory authorities does not ensure approval by regulatory authorities in other countries or by the FDA. The non-North American regulatory approval process may include all of the risks associated with obtaining FDA approval. We or a collaborator, if any, may not obtain non-North American regulatory approvals on a timely basis, if at all. We or a collaborator, if any, may not be able to file for non-North American regulatory approvals and may not receive necessary approvals to commercialize apatorsen in any market.
None.
We have business offices located in Bothell, Washington and Vancouver, British Columbia.
Our lease agreement for office space in Bothell, Washington commenced on February 15, 2015 and has a three-year term with one three-year renewal option. Pursuant to this lease, we rent approximately 13,771 square feet of office space. The annual rent is approximately $0.3 million.
We lease approximately 4,857 square feet in Vancouver, British Columbia, currently at an annual rent of approximately CND $0.1 million, which lease expires in September 2016.
We believe that the facilities we currently lease are sufficient for our anticipated near-term needs.
On January 5, 2016, Ionis Pharmaceuticals, Inc. (formerly known as Isis Pharmaceuticals, Inc.) filed a lawsuit against our subsidiary OncoGenex Technologies, Inc. in the United States District Court for the Southern District of California. Ionis claimed that OncoGenex Technologies was in breach of an Amended and Restated License Agreement between Ionis and OncoGenex Technologies dated July 2, 2008, as amended, or License Agreement. Under the License Agreement, Ionis is entitled to a share of certain forms of non-royalty revenue received by OncoGenex Technologies, but is not entitled to a share of revenue received by OncoGenex Technologies for the reimbursement of research and development activities. In April 2015, we terminated a collaboration agreement with Teva Pharmaceuticals Industries Ltd. In connection with that termination, Teva paid us $23.2 million as an advance reimbursement for certain continuing research and development activities related to custirsen and certain other antisense inhibitors of clusterin. In the lawsuit, Ionis claimed that OncoGenex Technologies was in breach of the License Agreement for failing to pay Ionis a share of the advance reimbursement payment from Teva and other non-monetary consideration received from Teva. Ionis sought damages and a declaratory judgment that, based on OncoGenex Technologies’ alleged breach, Ionis has the right to terminate the License Agreement. On March 4, 2016, OncoGenex Technologies filed a motion to dismiss the lawsuit in the United States District Court for the Southern District of California.
In August 2016, we and Ionis settled our lawsuit. Pursuant to the settlement, we paid to pay Ionis a $1.4 million upfront payment and were required to pay additional success-based payments up to an amount not exceeding $5.0 million. In November 2016, we provided a notice of discontinuance of custirsen to Ionis, the Notice of Discontinuance, and we believe that all financial obligations, other than continuing mutual indemnification obligations, under all agreements with Ionis, including the settlement agreement, are no longer owed and no further payments are due.
Not applicable.
32
|
MARKET FOR THE REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Our common stock first began trading on the Nasdaq National Market under the symbol “SNUS” on October 12, 1995. Following the completion of the Arrangement discussed elsewhere in this Annual Report on Form 10-K, our common stock commenced trading on the Nasdaq Capital Market under the stock symbol “OGXI”, effective August 21, 2008.
No cash dividends have been paid on our common stock, and we do not anticipate paying any cash dividends in the foreseeable future. As of February 15, 2017, there were approximately 57 stockholders of record and there were approximately 9,508 beneficial stockholders of our common stock. The high and low sales prices of our common stock as reported by the NASDAQ Capital Market for the periods indicated are as follows:
|
OncoGenex Pharmaceuticals, Inc. |
|
HIGH |
|
|
LOW |
|
||
|
|
|
|
|
|
|
|
|
|
|
YEAR ENDED DECEMBER 31, 2015: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
First quarter |
|
$ |
2.78 |
|
|
$ |
1.92 |
|
|
Second quarter |
|
|
3.10 |
|
|
|
1.74 |
|
|
Third quarter |
|
|
4.10 |
|
|
|
1.38 |
|
|
Fourth quarter |
|
|
2.80 |
|
|
|
1.11 |
|
|
|
|
|
|
|
|
|
|
|
|
YEAR ENDED DECEMBER 31, 2016: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
First quarter |
|
$ |
1.23 |
|
|
$ |
0.45 |
|
|
Second quarter |
|
|
1.42 |
|
|
|
0.68 |
|
|
Third quarter |
|
|
1.03 |
|
|
|
0.46 |
|
|
Fourth quarter |
|
|
0.70 |
|
|
|
0.33 |
|
The information required by this item regarding equity compensation plan information is set forth in Part III, Item 12 of this Annual Report on Form 10-K. No purchases of equity securities during the year ended December 31, 2016 were made by us or on our behalf and we did not sell any unregistered securities during such year.
33
The following performance graph shall not be deemed “filed” for purposes of Section 18 of the Exchange Act, or incorporated by reference into any of our filings under the Securities Act or the Exchange Act, except as expressly set forth by specific reference in such filings. The graph compares the cumulative five-year total return provided to stockholders on our common stock relative to the cumulative total returns of the NASDAQ Composite Index and the NASDAQ Pharmaceutical Index. An investment of $100 (with reinvestment of all dividends into additional shares of the same class of equity securities at the frequency with which dividends are paid on such securities during the applicable year) is assumed to have been made in our common stock and in each of the indexes on December 31, 2011 and its relative performance is tracked through December 31, 2016.
The stock price performance included in this graph is not necessarily indicative of future stock price performance.
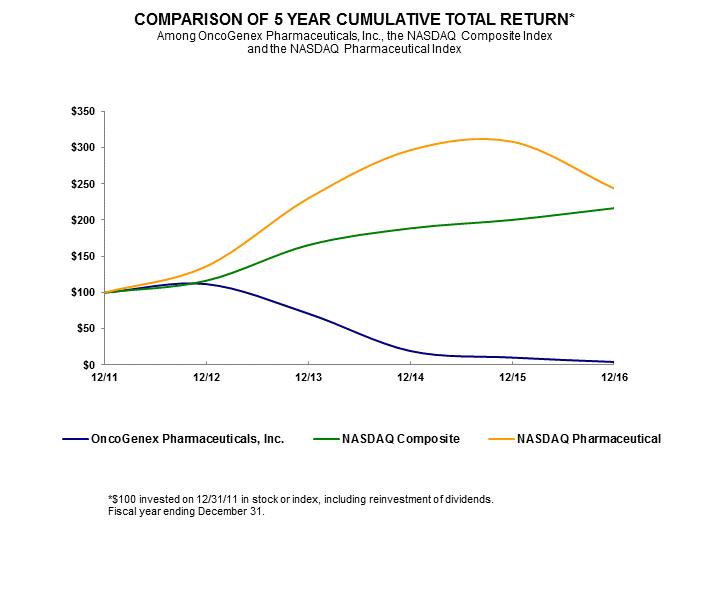
|
|
|
12/31/11 |
|
|
12/31/12 |
|
|
12/31/13 |
|
|
12/31/14 |
|
|
12/31/15 |
|
|
12/31/16 |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OncoGenex Pharmaceuticals, Inc. |
|
|
100.00 |
|
|
|
111.75 |
|
|
|
71.04 |
|
|
|
19.51 |
|
|
|
10.31 |
|
|
|
4.26 |
|
|
NASDAQ Composite |
|
|
100.00 |
|
|
|
116.41 |
|
|
|
165.47 |
|
|
|
188.69 |
|
|
|
200.32 |
|
|
|
216.54 |
|
|
NASDAQ Pharmaceutical |
|
|
100.00 |
|
|
|
136.13 |
|
|
|
229.92 |
|
|
|
296.47 |
|
|
|
308.15 |
|
|
|
243.63 |
|
34
The data set forth below should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the Consolidated Financial Statements and Notes thereto appearing at Item 8 of this Annual Report on Form 10-K. The selected consolidated statements of loss data for the years ended December 31, 2016, 2015 and 2014 and consolidated balance sheet data as of December 31, 2016 and 2015 set forth below have been derived from our audited consolidated financial statements included elsewhere in this Annual Report on Form 10-K. The selected statements of loss data for the year ended December 31, 2013 and 2012 and the balance sheet data as of December 31, 2014, 2013 and 2012 set forth below have been derived from the audited consolidated financial statements for such years not included in this Annual Report on Form 10-K.
The historical results presented are not necessarily indicative of future results.
|
|
|
December 31, |
|
|||||||||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||||
|
|
|
(in thousands except share and per share amounts) |
|
|||||||||||||||||
|
Statements of Loss Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Collaboration revenue |
|
$ |
5,062 |
|
|
$ |
18,160 |
|
|
$ |
27,116 |
|
|
$ |
29,882 |
|
|
$ |
20,095 |
|
|
Total expenses |
|
$ |
26,254 |
|
|
$ |
36,913 |
|
|
$ |
56,582 |
|
|
$ |
65,209 |
|
|
$ |
46,082 |
|
|
Net loss |
|
$ |
(20,129 |
) |
|
$ |
(16,801 |
) |
|
$ |
(26,240 |
) |
|
$ |
(31,849 |
) |
|
$ |
(21,098 |
) |
|
Basic and diluted loss per common share |
|
$ |
(0.67 |
) |
|
$ |
(0.64 |
) |
|
$ |
(1.45 |
) |
|
$ |
(2.17 |
) |
|
$ |
(1.56 |
) |
|
Shares used in calculation of net loss per share |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted |
|
|
29,949,432 |
|
|
|
26,147,344 |
|
|
|
18,098,799 |
|
|
|
14,683,389 |
|
|
|
13,522,723 |
|
|
|
|
December 31, |
|
|||||||||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|
2012 |
|
|||||
|
|
|
(in thousands) |
|
|||||||||||||||||
|
Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and short-term investments |
|
$ |
25,463 |
|
|
$ |
55,186 |
|
|
$ |
47,057 |
|
|
$ |
39,222 |
|
|
$ |
75,383 |
|
|
Total assets |
|
$ |
27,470 |
|
|
$ |
58,209 |
|
|
$ |
56,291 |
|
|
$ |
55,689 |
|
|
$ |
82,016 |
|
|
Current liabilities |
|
$ |
8,455 |
|
|
$ |
20,664 |
|
|
$ |
22,218 |
|
|
$ |
14,934 |
|
|
$ |
11,556 |
|
|
Total liabilities |
|
$ |
8,504 |
|
|
$ |
20,769 |
|
|
$ |
22,232 |
|
|
$ |
18,478 |
|
|
$ |
15,809 |
|
|
Additional paid-in capital |
|
$ |
213,239 |
|
|
$ |
211,590 |
|
|
$ |
191,373 |
|
|
$ |
168,242 |
|
|
$ |
165,395 |
|
|
Accumulated deficit |
|
$ |
(196,942 |
) |
|
$ |
(176,811 |
) |
|
$ |
(159,958 |
) |
|
$ |
(133,689 |
) |
|
$ |
(101,840 |
) |
|
Stockholders’ equity |
|
$ |
18,966 |
|
|
$ |
37,440 |
|
|
$ |
34,059 |
|
|
$ |
37,211 |
|
|
$ |
66,207 |
|
35
Forward-Looking Statements
This Annual Report on Form 10-K contains “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. These forward-looking statements involve a number of risks and uncertainties. We caution readers that any forward-looking statement is not a guarantee of future performance and that actual results could differ materially from those contained in the forward-looking statement. These statements are based on current expectations of future events. Such statements include, but are not limited to, statements about future financial and operating results, plans, objectives, expectations and intentions, costs and expenses, interest rates, outcome of contingencies, financial condition, results of operations, liquidity, business strategies, cost savings, objectives of management and other statements that are not historical facts. You can find many of these statements by looking for words like “believes,” “expects,” “anticipates,” “estimates,” “may,” “should,” “will,” “could,” “plan,” “intend,” or similar expressions in this Annual Report on Form 10-K or in documents incorporated by reference into this Annual Report on Form 10-K . We intend that such forward-looking statements be subject to the safe harbors created thereby. Examples of these forward-looking statements include, but are not limited to:
|
|
• |
the timing and completion of our pending merger; |
|
|
• |
our ability to identify a third party to develop apatorsen; |
|
|
• |
progress and preliminary and future results of clinical trial; |
|
|
• |
anticipated regulatory filings, requirements and future clinical trials; |
|
|
• |
timing and amount of future contractual payments, product revenue and operating expenses; |
|
|
• |
market acceptance of our products and the estimated potential size of these markets; and |
|
|
• |
our anticipated future capital requirements and the terms of any capital financing agreements. |
These forward-looking statements are based on the current beliefs and expectations of our management and are subject to significant risks and uncertainties. If underlying assumptions prove inaccurate or unknown risks or uncertainties materialize, actual results may differ materially from current expectations and projections. Factors that might cause such a difference include those discussed in Item 1A “Risk Factors,” as well as those discussed elsewhere in the Annual Report on Form 10-K.
You are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of this Annual Report on Form 10-K or, in the case of documents referred to or incorporated by reference, the date of those documents.
All subsequent written or oral forward-looking statements attributable to us or any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section. We do not undertake any obligation to release publicly any revisions to these forward-looking statements to reflect events or circumstances after the date of this Annual Report on Form 10-K or to reflect the occurrence of unanticipated events, except as may be required under applicable U.S. securities law. If we do update one or more forward-looking statements, no inference should be drawn that we will make additional updates with respect to those or other forward-looking statements.
Overview
We are a biopharmaceutical company that has been focused on the development of novel next generation cancer therapeutics. Our mission is to accelerate transformative therapies to improve the lives of people living with cancer and other serious diseases. Our product candidate apatorsen has a distinct mechanism of action and represents a unique opportunity for cancer drug development that we believe has the potential to improve treatment outcomes in a variety of cancers. Apatorsen is designed to block the production of heat shock protein 27, or Hsp27, a protein that promotes treatment resistance in cancer. In some clinical trials evaluating apatorsen, high serum Hsp27 levels appear to be a strong prognostic indicator for shorter survival outcomes. We currently do not intend to conduct additional pre-clinical or clinical studies with apatorsen and are seeking a collaboration partnership to fund and further develop this product candidate.
As a result of custirsen not meeting the primary endpoint of improving overall survival in three completed phase 3 trials, we have discontinued further development of custirsen and have begun to wind down all clinical trials and other activities related to this product candidate. In November 2016, we provided a notice of discontinuance to Ionis Pharmaceuticals, Inc. (formerly Isis Pharmaceuticals, Inc.), or Ionis, and a letter of termination to the University of British Columbia, or UBC, notifying those parties that we have discontinued development of custirsen, resulting in termination of all licensing agreements related to custirsen. In January 2017, we also discontinued further development of our pre-clinical product candidate, OGX-225. We provided a notice of discontinuance to Ionis,
36
informing them that we have discontinued development of OGX-225 resulting in termination of the license agreement related to this product candidate. We intend to also terminate the UBC license agreement related to OGX-225, provided that Ionis does not exercise its reversion rights within 90 days of the notice of discontinuance. If Ionis exercises its reversion rights related to OGX-225, we believe Ionis will assume the rights and obligations under the UBC license agreement.
In February 2016, we committed to a plan to reduce operating expenses, which included a workforce reduction of 11 employees, representing approximately 27% of our employees prior to the reduction. We incurred approximately $0.4 million in expenses as a result of the workforce reduction, substantially all of which were severance costs.
In October and November 2016, we committed to a restructuring of a portion of our workforce in order to preserve our resources as we determined future strategic plans. As part of these restructurings, we eliminated 19 positions, representing approximately 68% of our workforce. We expect the restructurings to be substantially complete in the first quarter of 2017. As of December 31, 2016, we incurred approximately $1.8 million in restructuring costs, substantially all of which related to severance costs, and an asset impairment charge of $0.2 million for manufacturing equipment.
On January 5, 2017, we and Achieve Life Science, Inc., or Achieve, a privately held specialty pharmaceutical company, entered into an Agreement and Plan of Merger and Reorganization, or the Merger Agreement, under which OncoGenex will acquire Achieve in an all-stock transaction. Upon completion of the Merger Agreement, Achieve's stockholders are expected to own 75% of the combined company's outstanding shares and our current equityholders are expected to own the remaining 25% of the combined company's outstanding shares. Following completion of the merger, OncoGenex Pharmaceuticals, Inc. will be renamed Achieve Life Sciences, Inc.
Pending Merger Agreement with Achieve
On January 5, 2017, we and Achieve entered into the Merger Agreement, pursuant to which Ash Acquisition Sub, Inc., a Delaware corporation and a wholly owned subsidiary of ours will merge with and into Achieve, or the First Merger, with Achieve becoming a wholly owned subsidiary of ours and the surviving company of the First Merger, or the Initial Surviving Corporation. Promptly following the First Merger, the Initial Surviving Corporation will merge with and into Ash Acquisition Sub 2, Inc., or Merger Sub 2, a Delaware corporation and a wholly owned subsidiary of ours, with Merger Sub 2 continuing as the surviving entity as a direct wholly owned subsidiary of ours. The two mergers taken together, are intended to qualify as a “reorganization” within the meaning of Section 368(a)(2)(D) of the Internal Revenue Code of 1986, as amended. The surviving company is expected to be renamed Achieve Life Sciences, Inc. and is referred to herein as the “combined company.” The Merger is expected to close mid-2017.
Subject to the terms and conditions of the Merger Agreement, at the closing of the First Merger, each outstanding share of Achieve common stock will be converted into the right to receive approximately 4,242.8904 shares of our common stock, subject to adjustment as provided in the Merger Agreement based on increases or decreases in Achieve’s fully-diluted capitalization, as well as the payment of cash in lieu of fractional shares. Immediately following the effective time of the merger, our equityholders are expected to own approximately 25% of the outstanding capital stock of the combined company on a fully diluted basis, and the Achieve stockholders are expected to own approximately 75% of the outstanding capital stock of the combined company on a fully diluted basis.
Consummation of the merger is subject to certain closing conditions, including, among other things, approval by the stockholders of us and Achieve. The Merger Agreement contains certain termination rights for both us and Achieve, and further provides that, upon termination of the Merger Agreement under specified circumstances, either party may be required to pay the other party a termination fee of $0.5 million. In addition, the Merger Agreement provides that if either party breaches certain covenants regarding alternative transactions to those contemplated by the Merger Agreement, the breaching party may be required to pay the other party a termination fee of $1.0 million. In connection with certain terminations of the Merger Agreement, either party may be required to pay the other party’s third party expenses up to $0.5 million.
At the effective time of the First Merger, our Board of Directors is expected to consist of seven members, three of whom will be designated by us and four of whom will be designated by Achieve. We are expected to designate Scott Cormack, Stewart Parker and Martin Mattingly. Achieve is expected to designate Richard Stewart, Anthony Clark and two other independent directors that have yet to be determined. Additionally, at the effective time of the First Merger, Rick Stewart, the current Chairman of Achieve, is expected to be the Chairman and Chief Executive Officer of the combined company; Anthony Clarke, the current Chief Scientific Officer of Achieve, is expected to be the Chief Scientific Officer of the combined company; and John Bencich, our Chief Financial Officer and Cindy Jacobs, our Chief Medical Officer, are expected to continue to serve the combined company in their respective roles.
In accordance with the terms of the Merger Agreement, (i) certain of our officers and directors, who collectively hold approximately 1.2 percent of the outstanding shares of our capital stock as of the close of business on January 4, 2017, have each entered into a support agreement with Achieve, or the OncoGenex Support Agreements, and (ii) certain officers, directors and stockholders of
37
Achieve, who collectively hold approximately 78 percent of the outstanding shares of Achieve capital stock as of the close of business on January 4, 2017, have each entered into a support agreement with us, or the Achieve Support Agreements, and together with the OncoGenex Support Agreements, the Support Agreements. The Support Agreements include covenants as to the voting of such shares in favor of approving the transactions contemplated by the Merger Agreement and against actions that could adversely affect the consummation of the Merger.
The Support Agreements will terminate upon the earlier of the consummation of the First Merger or the termination of the Merger Agreement by its terms.
Concurrently and in connection with the execution of the Merger Agreement, (i) certain of our officers and directors, who collectively hold approximately 1.2 percent of the outstanding shares of our capital stock as of the close of business on January 4, 2017 and (ii) certain officers, directors and stockholders of Achieve, who collectively hold approximately 78 percent of the outstanding shares of Achieve capital stock as of the close of business on January 4, 2017, have each entered into lock-up agreements with us, pursuant to which, subject to certain exceptions, each stockholder will be subject to a 180-day, or the Lock-Up Period, lock-up on the sale of shares of our capital stock, which Lock-Up Period shall begin upon the consummation of the First Merger.
We expect to issue contingent value rights, or each, a CVR and collectively, the CVRs, to our existing stockholders prior to the completion of the First Merger. One CVR will be issued for each share of our common stock outstanding as of the record date for such issuance. Each CVR will be a non-transferable right to potentially receive certain cash, equity or other consideration received by the combined company in the event the combined company receives any such consideration during the five-year period after consummation of the First Merger as a result of the achievement of certain clinical milestones, regulatory milestones, sales-based milestones and/or up-front payment milestones relating to our product candidate apatorsen, or the Milestones, upon the terms and subject to the conditions set forth in a contingent value rights agreement to be entered into between us, Achieve and an as of yet unidentified third party, as rights agent, or the CVR Agreement. The aggregate consideration to be distributed to the holders of the CVRs, if any, will be equal to 80% of the consideration received by the combined company as a result of the achievement of the Milestones less certain agreed to offsets, as determined pursuant to the CVR Agreement. Under the CVR Agreement, for a period of six months beginning in February 2017, we will use certain defined efforts to enter into an agreement with a third party regarding the development and/or commercialization of apatorsen. At the expiration of this six-month period, if a third party has not entered into a term sheet for the development or commercialization of apatorsen, the combined company will no longer be contractually required to pursue an agreement regarding apatorsen and no consideration will be payable to the holders of CVRs.
We also entered into a letter agreement with Achieve, whereby we would pay, on behalf of Achieve, for transactions costs associated with the merger. In the event that the Merger Agreement is terminated and as a result of such termination we are required to pay to Achieve one or more termination fees, the total amount of termination fees we would owe is reduced by the amount of the transaction costs we would have paid on behalf of Achieve.
Product Candidate Apatorsen
Apatorsen is our product candidate that is designed to inhibit production of Hsp27, a cell-survival protein expressed in many types of cancers including bladder, prostate, breast, pancreatic and non-small cell lung cancer. Hsp27 expression is stress-induced, including by many anti-cancer therapies. Overexpression of Hsp27 is thought to be an important factor leading to the development of treatment resistance and is associated with metastasis and negative clinical outcomes in patients with various tumor types. In some clinical trials evaluating apatorsen, high serum Hsp27 levels at baseline, or at the start of treatment, appear to be a strong prognostic indicator for shorter survival outcomes.
Apatorsen utilizes second-generation antisense drug chemistry and belongs to the drug class known as antisense therapeutics. We have collaborated with Ionis and selectively licensed technology from Ionis to combine Ionis’ second-generation antisense chemistry with our proprietary gene target sequences to create an inhibitor that is designed to down-regulate Hsp27. In contrast to first-generation antisense chemistry, second-generation antisense chemistry has improved target binding affinity, increased resistance to degradation and improved tissue distribution. These improvements result in slower clearance of the therapies from the body, which allow for less frequent dosing and thereby make treatment easier on patients at a lower associated cost.
A number of preclinical studies have shown that reducing Hsp27 production induces tumor cell death in prostate, non-small cell lung, bladder and pancreatic cancer cells. The studies also suggest that reducing Hsp27 production sensitizes prostate tumor cells to hormone ablation therapy. These preclinical studies have also shown that inhibiting the production of Hsp27 in human prostate, bladder, lung, breast, ovarian and pancreatic tumor cells sensitizes the cells to chemotherapy.
Hsp27 has been reported by others to function as an immunomodulatory protein by a number of mechanisms that include altering important membrane-expressed proteins on monocytes and immature dendritic cells; this alteration results in tumor-associated
38
immune cells that are not functional in identifying and killing cancer cells. The induction of anti-inflammatory cytokines by Hsp27 may also play a role in down-regulating lymphocyte activation leading to additional unresponsive immune cells.
In 2013, we initiated the ORCA (Ongoing Studies Evaluating Treatment Resistance in CAncer) program which encompasses six phase 2 clinical studies designed to evaluate whether treatment with apatorsen can lead to improved prognosis and treatment outcomes for cancer patients. Five of these trials have been completed and the remaining ongoing trial completed enrollment in 2016 with results expected in 2018. We currently do not intend to conduct additional pre-clinical or clinical studies with apatorsen and are seeking a collaboration partnership to fund and further develop this product candidate.
Six phase 2 apatorsen clinical trials have been initiated or completed under the ORCA program.
Ongoing Trial
|
|
• |
The Spruce-2™ Trial (formerly referred to as the Cedar Trial): The completed investigator-sponsored, randomized phase 2 trial evaluating apatorsen plus gemcitabine and carboplatin therapy or gemcitabine and carboplatin therapy alone in patients with previously untreated advanced squamous non-small cell lung cancer, or NSCLC. Patients also continue weekly apatorsen infusions as maintenance treatment after chemotherapy until disease progression. The aim of the trial is to determine if adding apatorsen to gemcitabine and carboplatin therapy can extend progression free survival, or PFS, outcome. Additional analyses will include tumor response rates, overall survival, safety, and health-related quality of life, as well as to determine the effect of Hsp27 levels on clinical outcomes, explore potential biomarkers that may help predict response to treatment and survival outcomes in patients who were at increased risk for poor outcomes. The trial was initiated in July 2014 and completed enrollment in December 2016. During the conduct of the trial, two amendments were submitted: one that reduced the apatorsen dose to 400mg and the second that reduced patient enrollment to ~90 patients. The trial completed patient enrollment in December 2016 and results are expected in the second half of 2017. The trial is an investigator-sponsored trial being conducted and funded primarily by the UK National Cancer Research Network and the UK Experimental Cancer Medicine Network. |
Completed Trials
|
|
• |
The Borealis-2™ Trial: The completed investigator-sponsored, randomized phase 2 trial evaluated apatorsen in combination with docetaxel treatment compared to docetaxel treatment alone in patients with advanced or metastatic bladder cancer who have disease progression following first-line platinum-based chemotherapy. The primary endpoint analysis was a superiority test for overall survival, performed at a one-sided 0.10 significance level using a stratified log-rank test. Secondary endpoints included PFS, disease response and safety assessments. The trial met its primary endpoint of improving survival at the one-sided 0.10 significance level. Patients who received apatorsen treatment experienced a 20% reduction in risk of death, compared to patients receiving docetaxel alone (overall survival hazard ratio (HR)=0.80; 80% CI: 0.65-0.98; p=0.078). In February 2017, results were presented at the American Society of Clinical Oncology, or ASCO, 2017 Genitourinary Cancers Symposium. Apatorsen was well tolerated in combination with docetaxel. The reduction in risk of progression or death was 20% for patients receiving apatorsen in combination with docetaxel, compared to docetaxel alone (PFS HR= 0.80; 80% CI: 0.64-1.01; p=0.107). Partial or complete responses occurred in 16.2% patients receiving apatorsen plus docetaxel compared to 10.9% patients receiving docetaxel alone with median response durations of 6.2 months versus 4.4 months, respectively. Overall for the study, higher baseline serum Hsp27 levels were significantly prognostic for indicating an almost 2-fold higher risk of death (HR= 1.96; p=0.0001). In an exploratory analysis on a subset of patients (20% of total) who completed at least 2 treatment cycles and had either a decrease in serum Hsp27 levels from baseline or had only a 20.5% increase in serum Hsp27 levels from baseline, the reduction in risk of death with apatorsen treatment was 71% (HR= 0.29: 80% CI: 0.18-0.48; interaction p=0.0727). The trial was conducted by the Hoosier Cancer Research Network at 28 sites across the United States. |
39
|
|
• |
The Spruce™ Trial: The investigator-sponsored, randomized, placebo-controlled phase 2 trial evaluating apatorsen plus carboplatin and pemetrexed therapy compared to carboplatin and pemetrexed therapy in patients with previously untreated advanced non-squamous NSCLC. Patients continued pemetrexed with weekly apatorsen or placebo infusions as maintenance treatment until disease progression if they completed a minimum of 3 cycles of chemotherapy treatment. The aim of the trial is to determine if adding apatorsen to carboplatin and pemetrexed therapy could extend PFS outcome. In January 2016, the primary endpoint data for PFS was reported to have not reached the statistical significance required to demonstrate a benefit (PFS HR= 0.90; 80% CI 0.71-1.14; p=0.557). In the study, higher baseline serum Hsp27 levels were found to be significantly prognostic for indicating an almost 2-fold higher risk of death (HR= 1.98; p=0.0034). A potential benefit was observed in a subgroup of patients with high baseline serum Hsp27 status (~10% of total) when treated with apatorsen (PFS HR= 0.462; 80% CI: 0.193- 1.106). Study follow up with survival results was completed at the end of 2016. The addition of apatorsen to carboplatin and pemetrexed therapy did not demonstrate an overall survival benefit in the study (HR= 1.067; 80% CI: 0.838-1.359). PFS results were presented at ASCO 2016. The study investigators concluded that apatorsen and pemetrexed/carboplatin therapy was well tolerated and showed promising PFS results in the treatment of patients with non-squamous NSCLC who have Hsp27 high status and thus warranted further study in this population. We do not intend to pursue additional trials in non-squamous NSCLC at this time. The study was an investigator-sponsored trial conducted by sites under the Sarah Cannon Research Institute. |
|
|
• |
The Rainier™ Trial: Our completed investigator-sponsored Rainier™ phase 2 trial was a randomized, placebo-controlled trial evaluating apatorsen in combination with ABRAXANE® (paclitaxel protein-bound particles for injectable suspension) (albumin-bound) and gemcitabine compared to ABRAXANE and gemcitabine alone in patients with untreated metastatic pancreatic cancer. The addition of apatorsen to ABRAXANE and gemcitabine did not show improved survival for patients receiving apatorsen plus ABRAXANE and gemcitabine when compared to ABRAXANE and gemcitabine alone (HR= 1.098; 95% CI 0.759-1.590). Similarly there was no improvement in PFS (PFS HR=1.020; 95% CI 0.806-1.290). The study did show that higher baseline serum Hsp27 levels were significantly prognostic for indicating a 1.8-fold higher risk of death (HR= 1.84; p=0.0041). A potential benefit was observed in a subgroup of patients with high baseline serum Hsp27 status (14% of total) when treated with apatorsen (PFS HR= 0.381; 95% CI 0.120-1.208 and survival HR= 0.587; 95% CI 0.195-1.770). The study was presented at the Gastrointestinal, or GI, Cancers Symposium meeting in January 2016. The study investigators concluded that these promising results in pancreatic cancer patients with high baseline Hsp27 status warrant further study of apatorsen in this population. We do not intend to pursue additional trials in pancreatic cancer at this time. The study was an investigator-sponsored trial conducted by sites under the Sarah Cannon Research Institute. |
40
Product Candidate Custirsen
In November 2016, we provided a notice of discontinuance to Ionis notifying them that we have discontinued development of custirsen, resulting in termination of all licensing agreements related to custirsen. We believe that all financial obligations, other than continuing mutual indemnification obligations and our requirement to pay for out-of-pocket patent expenses incurred up to the date of termination and for abandoning the custirsen patents and patent applications, under all agreements with Ionis, including the Ionis settlement agreement, are no longer owed and no further payments are due.
Product Candidate OGX-225
In January 2017, we discontinued further development of OGX-225. We provided a notice of discontinuance to Ionis, notifying them that we have discontinued development of OGX-225 resulting in termination of the license agreement related to this product candidate. We intend to also terminate the UBC license agreement related to OGX-225 provided that Ionis does not exercise its reversion rights within 90 days of the notice of discontinuance. If Ionis exercises its reversion rights related to OGX-225, we believe Ionis will assume the rights and obligations under the UBC license agreement.
Collaboration Revenue
Revenue recognized to date was attributable to the upfront payment we received in the fourth quarter of 2009 pursuant to a Collaboration Agreement with Teva, as well as cash reimbursements from Teva for certain costs incurred by us under the clinical development plan. Our policy is to account for these reimbursements as collaboration revenue.
In April 2015, we and Teva entered into an agreement to terminate the Collaboration Agreement, or the Termination Agreement. Pursuant to the Termination Agreement, Teva paid to us, as advanced reimbursement for certain continuing research and development activities related to custirsen and certain other antisense inhibitors of clusterin, an amount equal to $27.0 million, less approximately $3.8 million that Teva paid for custirsen development on our behalf. We do not expect to receive any additional amounts from Teva. Teva is responsible for expenses related to custirsen incurred pursuant to the Collaboration Agreement through December 31, 2014. We are responsible for certain custirsen-related expenses from and after January 1, 2015.
As a result of the termination of the Collaboration Agreement with Teva, we do not expect to earn any additional collaboration revenue beyond the amounts provided as advanced reimbursement for custirsen -related development expenses as set forth in the Termination Agreement. The advanced reimbursement payment made by Teva, as part of the Termination Agreement, was deferred and was recognized as collaboration revenue on a dollar for dollar basis as costs were incurred as part the of continuing research and development activities related to custirsen and certain other antisense inhibitors of clusterin. We have fully utilized the $23.2 million in advance reimbursement for custirsen-related development costs between January 1, 2015 and June 30, 2016.
Research and Development Expenses
Research and development, or R&D, expenses consist primarily of costs for clinical trials, contract manufacturing, personnel costs, milestone payments to third parties, facilities, regulatory activities, preclinical studies and allocations of other R&D-related costs. External expenses for clinical trials include fees paid to clinical research organizations, clinical trial site costs and patient treatment costs.
41
Currently, we manage our clinical trials through contract research organizations and independent medical investigators at their sites and at hospitals and expect this practice to continue. Due to the number of projects and our ability to utilize resources across several projects, we do not record or maintain information regarding the indirect operating costs incurred for our research and development programs on a program-specific basis. In addition, we believe that allocating costs on the basis of time incurred by our employees does not accurately reflect the actual costs of a project.
Several of our clinical trials have been supported by grant funding that was received directly by the hospitals and/or clinical investigators conducting the clinical trials as investigator-sponsored trials, thereby allowing us to complete these clinical trials at a lower cost to us.
In accordance with the Termination Agreement, Teva was required to and did fund all additional expenses under the clinical development plan through December 31, 2014, after which date we took over responsibility for future custirsen-related costs following termination of our Collaboration Agreement. We do not owe Teva any development milestone payments or royalty payments on sales of custirsen, if any.
We cannot estimate completion dates for development activities or when we might receive material net cash inflows from our R&D projects, if ever.
Our projects or intended R&D activities may be subject to change from time to time as we evaluate results from completed studies, our R&D priorities and available resources.
General and Administrative Expenses
General and administrative, or G&A, expenses consist primarily of salaries and related costs for our personnel in executive, finance and accounting, corporate communications, human resources and other administrative functions, as well as consulting costs, including market research, business consulting and intellectual property. Other costs include professional fees for legal and auditing services, insurance and facility costs.
Warrant liability
The following is a summary of outstanding warrants to purchase common stock that are classified as liabilities at December 31, 2016:
|
|
|
Total |
|
|
|
|
|
|
|
|
|
|
|
Outstanding |
|
|
Exercise |
|
|
|
||
|
|
|
and |
|
|
price per |
|
|
|
||
|
|
|
Exercisable |
|
|
Share |
|
|
Expiration Date |
||
|
(1) Series A Warrants issued in July 2014 financing |
|
|
2,779,933 |
|
|
|
4.00 |
|
|
July 2019 |
|
(2) Series B Warrants issued in July 2014 financing |
|
|
670,269 |
|
|
|
4.00 |
|
|
July 2019 |
No warrants classified as liabilities were exercised during the years ended December 31, 2016 or 2015.
We reassess the fair value of the common stock warrants classified as liabilities at each reporting date utilizing a Black-Scholes pricing model. Inputs used in the pricing model include estimates of stock price volatility, expected warrant life and risk-free interest rate. The computation of expected volatility was based on the historical volatility of shares of our common stock for a period that coincides with the expected life of the warrants.
Results of Operations
Years Ended December 31, 2016, 2015 and 2014
Revenue
Revenue for the years ended December 31, 2016, 2015 and 2014 were $5.1 million, $18.2 million and $27.1 million, respectively. The advanced reimbursement payment made by Teva, as part of the Termination Agreement, was deferred and recognized as collaboration revenue on a dollar for dollar basis as costs were incurred as part of the continuing research and development activities related to custirsen. The decrease in collaboration revenue in 2016 as compared to 2015 was due to the full recognition of the remaining amounts of deferred revenue in 2016. The decrease in 2015 as compared to 2014 was due primarily to lower collaboration revenue recognized for the reimbursement of expenses for the phase 3 clinical trial in second-line chemotherapy in patients with metastatic CRPC, or the AFFINITY trial, as a result of patients coming off treatment. This was partially offset by higher trial costs in the phase 3
42
clinical trial in second-line chemotherapy in patients with NSCLC, or the ENSPIRIT trial, which OncoGenex became responsible for pursuant to the Termination Agreement with Teva. Revenue recognized in 2015 is attributable to the advance reimbursement received in the second quarter of 2015, pursuant to the Termination Agreement with Teva, for research and development costs incurred by us related to the custirsen development program.
Research and Development Expenses
Our research and development expenses for our clinical development programs were as follows (in thousands):
|
|
|
Year ended |
|
|||||||||
|
|
|
December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Clinical development programs: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Custirsen |
|
$ |
8,959 |
|
|
$ |
15,544 |
|
|
$ |
26,015 |
|
|
Apatorsen |
|
$ |
1,521 |
|
|
$ |
2,798 |
|
|
$ |
9,753 |
|
|
Other research and development |
|
$ |
4,308 |
|
|
$ |
6,766 |
|
|
$ |
10,456 |
|
|
Total research and development expenses |
|
$ |
14,788 |
|
|
$ |
25,108 |
|
|
$ |
46,224 |
|
Research and development expenses for the years ended December 31, 2016, 2015 and 2014 were $14.8 million, $25.1 million and $46.2 million, respectively. The decrease in 2016 as compared to 2015 was due to lower clinical trial costs for the AFFINITY trial, the ENSPIRIT trial and our investigator sponsored apatorsen trials, lower consulting and professional fees as a result of the restructuring in the first and fourth quarters of 2016 and a decrease in facilities costs. The decrease in 2015 as compared to 2014 was due primarily to lower clinical trial costs for Borealis-1 as a result of patients coming off treatment and fewer combination drug purchases for the AFFINITY trial in 2014.
General and Administrative Expenses
G&A expenses for the years ended December 31, 2016, 2015 and 2014 were $8.9 million, $11.8 million and $10.6 million, respectively. The decrease in 2016 as compared to 2015 was due to lower professional fees and headcount and consulting expenses as a result of the restructurings in the first and fourth quarters of 2016. The increase in 2015 as compared to 2014 was primarily due to higher consulting and legal fees. This was partially offset by lower rent and facilities operating costs and lower employee related costs.
Revaluation of Warrants
We recorded gains on the revaluation of our outstanding warrants for the years ended December 31, 2016, 2015 and 2014 of $0.9 million, $1.9 million and $3.7 million, respectively, which is included on our consolidated statement of loss as a gain on warrants. We revalue the warrants at each balance sheet date to fair value.
Restructuring recovery / (expense)
We recorded a restructuring expense of $2.2 million for the year ended December 31, 2016 and a restructuring recovery of $0.3 million for the year ended December 31, 2014.
In February 2016, we committed to a plan to reduce operating expenses, which included a workforce reduction of 11 employees, representing approximately 27% of our employees prior to the reduction. We incurred approximately $0.4 million in expenses as a result of the workforce reduction, substantially all of which were severance costs.
In October 2016, we committed to a restructuring of a portion of our workforce in order to preserve our resources as we determine future strategic plans. As part of this restructuring, we eliminated 14 positions, representing approximately 48% of our workforce. We expect the restructuring to be substantially complete in the first quarter of 2017. As of December 31, 2016, we incurred approximately $1.1 million in restructuring costs, substantially all of which related to severance costs.
In November 2016, we committed to a further reduction in our workforce. We eliminated five positions and incurred approximately $0.7 million in expenses as a result of the workforce reduction, substantially all of which were severance costs.
|
|
|
Total |
|
|
Amounts |
|
|
Accrued at |
|
|||
|
|
|
estimated |
|
|
settled |
|
|
December 31, |
|
|||
|
|
|
costs |
|
|
to date |
|
|
2016 |
|
|||
|
Restructuring Costs |
|
$ |
2,206 |
|
|
$ |
(708 |
) |
|
$ |
1,498 |
|
43
We entered into a lease termination agreement with BMR-217TH Place LLC effective 2014 in relation to our previous Bothell facility. The termination of the lease resulted in a $3.5 million restructuring gain recorded in the fourth quarter of 2014, which was partially offset by a $3.2 million termination fee recognized in the same period.
Recovery of lease termination loss
We recorded a recovery of lease termination loss of $1.3 million for the year ended December 31, 2016. In February 2015, we entered into a Lease Termination Agreement with BMR pursuant to which we and BMR agreed to terminate our prior lease, effective March 1, 2015. Under the Lease Termination Agreement, we paid BMR a $2.0 million termination fee and would have been required to pay an additional $1.3 million termination fee if we had (i) met the primary endpoint for our AFFINITY Trial and if we had (ii) closed a transaction or transactions pursuant to which we received funding in an aggregate amount of at least $20.0 million. As of December 31, 2014 and subsequent annual and interim reporting periods up to June 30, 2016, we had assessed that the likelihood of meeting both contingent events was probable and as a result, recognized the $1.3 million in lease termination liability on our balance sheet as at the end of those reporting periods. In August 2016, final survival results of our AFFINITY trial did not meet the primary endpoint of a statistically significant improvement in overall survival in men with metastatic CRPC. As at September 30, 2016, we re-assessed that the likelihood of meeting both contingent events is no longer possible due to not achieving the primary endpoint on our AFFINITY trial. As a result, we have reversed the $1.3 million in lease termination liability on our balance sheet and recognized a recovery on our statement of loss.
Litigation settlement expense
In August 2016, we and Ionis settled our lawsuit. Pursuant to the settlement, we paid to pay Ionis a $1.4 million upfront payment and were required to pay additional success-based payments up to an amount not exceeding $5.0 million. In accordance with the upfront payment, we recorded litigation settlement expense of $1.4 million for the year ended December 31, 2016. In November 2016, we provided the Notice of Discontinuance to Ionis and we believe that all financial obligations, other than continuing mutual indemnification obligations, under all agreements with Ionis, including the settlement agreement, are no longer owed and no further payments are due.
Asset impairment charge
In the fourth quarter of 2016, we concluded that we had a triggering event requiring assessment of impairment for certain of our long-lived assets in conjunction with our restructuring actions announced in October 2016. As a result, we reviewed our long-lived assets for impairment and recorded a $0.2 million impairment charge, representing the entire amount of the then carrying value of the assets, on our statement of loss. The full amount of the impairment charge related to drug product manufacturing equipment.
Liquidity and Capital Resources
We have incurred an accumulated deficit of $196.9 million through December 31, 2016, and we expect to incur substantial additional losses in the future. We have not generated any revenue from product sales to date, and we may not generate product sales revenue in the near future, if ever.
Our operations to date have been primarily funded through the sale of our equity securities and payments received from Teva. As of December 31, 2016, our cash, cash equivalents, and short-term investments decreased to $25.5 million as compared to $55.2 million as of December 31, 2015.
In April 2015, we and Teva terminated our Collaboration Agreement. Pursuant to the Termination Agreement, Teva paid to us, as advanced reimbursement for certain continuing research and development activities related to custirsen, an amount equal to $23.2 million. We do not expect to receive any additional amounts from Teva. We fully utilized the $23.2 million in advance reimbursement for custirsen-related development costs between January 1, 2015 and June 30, 2016. We do not owe Teva any development milestone payments or royalty payments on sales of custirsen, if any.
In April 2015, we and Lincoln Park Capital Fund, LLC, or LPC, entered into a Purchase Agreement, pursuant to which we had the right to sell to LPC up to $18.0 million in shares of our common stock, subject to certain limitations and conditions set forth in the Purchase Agreement. LPC initially purchased 956,938 Series A-1 Units at a purchase price of $2.09 per unit, for aggregate gross proceeds of $2.0 million. Each Series A-1 Unit consisted of (i) one share of common stock and (ii) one warrant to purchase one-quarter of a share of common stock at an exercise price of $2.40 per share.
44
From April 30, 2015 through August 13, 2015, we offered and sold 6,814,980 shares of our common stock pursuant to our Purchase Agreement with LPC. These sales resulted in gross proceeds to us of approximately $18.0 million and offering expenses of $0.4 million. As of August 13, 2015, no further amounts remained available for sale under this offering program.
In February 2016, we committed to a plan to reduce operating expenses, which included a workforce reduction of 11 employees, representing approximately 27% of our employees prior to the reduction. We incurred approximately $0.4 million in expenses as a result of the workforce reduction, substantially all of which were severance costs.
In October 2016, we committed to a restructuring of a portion of our workforce in order to preserve our resources as we determine future strategic plans. As part of this restructuring, we eliminated 14 positions, representing approximately 48% of our workforce. We expect the restructuring to be substantially complete in the first quarter of 2017. As of December 31, 2016, we incurred approximately $1.1 million in restructuring costs, substantially all of which related to severance costs and an asset impairment charge of $0.2 million related to manufacturing equipment.
In November 2016, we committed to a further reduction in our workforce. We eliminated five positions and incurred approximately $0.7 million in expenses as a result of the workforce reduction, substantially all of which were severance costs. The workforce reduction was substantially completed in the fourth quarter of 2016.
Cash Flows
Operating Activities
For the years ended December 31, 2016, 2015 and 2014, net cash used in operating activities was $29.7 million, $9.1 million, and $17.3 million, respectively. The increase in cash used in operations in 2016 as compared to cash provided by operations in 2015 was primarily attributable to the advanced reimbursement payment made by Teva, as part of the Termination Agreement, which was received in the second quarter of 2015. The decrease in cash used in operations in 2015 as compared to cash used for operations in 2014 was primarily attributable to a cash payment from Teva as an advance reimbursement for custirsen development costs associated with the Termination Agreement in 2015.
Financing Activities
For the year ended December 31, 2016, net cash used by financing activities was $2,000, compared to net cash provided by financing activities of $17.6 million and $25.2 million for the years ended December 31, 2015 and 2014, respectively. Net cash used in financing activities for the year ended December 31, 2016 relates to taxes paid on the net share settlement of equity awards. Net cash provided by financing activities for the year ended December 31, 2015 relates to proceeds received from the financing through our purchase agreement with LPC. Net cash provided by financing activities for the year ended December 31, 2014 was the result of proceeds from the underwritten registered direct offering completed in July 2014, the sale of shares of common stock through our “at the market” equity offering program and the exercise of stock options.
Investing Activities
Net cash provided by investing activities for the year ended December 31, 2016 was $10.6 million, compared to net cash used in investing activities for the year ended December 31, 2015 of $2.1 million and net cash provided by investing activities for the year ended December 31, 2014 of $5.4 million. Net cash used in and provided by investing activities in all years was due to transactions involving marketable securities in the normal course of business.
Operating Capital and Capital Expenditure Requirements
Based on our current expectations, we believe that our cash, cash equivalents, and short-term investments will be sufficient to fund our currently planned operations for at least the next 12 months.
We have based this estimate on assumptions that may prove to be wrong, or we could utilize our available capital resources sooner than we currently expect. If the timeline to complete the recently announced merger takes longer than anticipated or is not completed, we change our development plans or elect to further develop apatorsen, cannot find third-party collaborators to fund further development of apatorsen, our ongoing trial proceeds slower or takes longer than expected to complete, we acquire rights to new product candidates, do not successfully defend litigation or engage in commercialization and product launch activities, we will need additional capital sooner than we expect. If we need to extend our cash availability or to conduct any such currently unplanned development activities, we would seek such necessary funding through the licensing or sale of our product candidate, by executing a
45
partnership or collaboration agreement, or through private or public offerings of our equity or debt. However, we can provide no assurance that such funding would be available to us on favorable terms, or at all.
Our future capital requirements will depend on many factors, including:
|
|
• |
the timing of completion of the pending merger with Achieve; |
|
|
• |
whether we modify our development program for apatorsen, including terminating and starting new trials; |
|
|
• |
whether we are able to enter into additional third-party collaborative partnerships to develop and/or commercialize apatorsen on terms that are acceptable to us; |
|
|
• |
the scope and results of our clinical trials and preclinical studies; |
|
|
• |
our ability to forecast the cost of our ongoing development activities; |
|
|
• |
whether we experience delays in our development program of apatorsen, or experience slower-than-anticipated product development or rate of events; |
|
|
• |
conducting studies required to obtain regulatory approvals for apatorsen from regulatory agencies; |
|
|
• |
the availability of third parties to perform the key development tasks for apatorsen, including conducting preclinical studies and clinical trials and manufacturing apatorsen to be tested in those studies and trials and the associated costs of those services; |
|
|
• |
the costs involved in preparing, filing, prosecuting, maintaining, defending the validity of and enforcing patent claims and other costs related to patent rights and other intellectual property rights, including litigation costs and the results of such litigation; |
|
|
• |
whether opportunities to acquire additional product candidates arise and the costs of acquiring and developing those product candidates; |
|
|
• |
the costs to defend, and the results of, litigation; and |
|
|
• |
whether we engage in commercialization and product launch activities. |
Contractual Obligations
The following table summarizes our contractual obligations as of December 31, 2016 (in thousands):
|
|
|
Total |
|
|
Less than 1 year |
|
|
1-3 years |
|
|
3-5 years |
|
|
More than 5 years |
|
|||||
|
Bothell office operating lease (1) |
|
$ |
376 |
|
|
$ |
281 |
|
|
$ |
95 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Vancouver office operating lease (2) |
|
$ |
68 |
|
|
$ |
68 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
UBC license maintenance fees (3) |
|
$ |
26 |
|
|
$ |
4 |
|
|
$ |
9 |
|
|
$ |
9 |
|
|
$ |
4 |
|
|
Leased equipment |
|
$ |
22 |
|
|
$ |
19 |
|
|
$ |
3 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Total |
|
$ |
492 |
|
|
$ |
372 |
|
|
$ |
107 |
|
|
$ |
9 |
|
|
$ |
4 |
|
|
(1) |
This operating lease is effective May 1, 2015 and expires on April 30, 2018. |
|
(2) |
This operating lease expires in 2017. |
|
(3) |
We are obligated to pay an annual license maintenance fee of CAD$6,000 to UBC, which has been converted to US dollars based on the December 31, 2016 exchange rate of US$1.00 = CAD$1.34551, and rounded to the nearest $1,000. |
Off-Balance Sheet Arrangements
We do not have any off-balance sheet financing arrangements at December 31, 2016.
Inflation
We do not believe that inflation has had a material effect on our business and results of operations during the periods presented.
46
Material Changes in Financial Condition
|
|
|
December 31, |
|
|||||
|
(in thousands) |
|
2016 |
|
|
2015 |
|
||
|
Total Assets |
|
$ |
27,470 |
|
|
$ |
58,209 |
|
|
Total Liabilities |
|
|
8,504 |
|
|
|
20,769 |
|
|
Total Equity |
|
|
18,966 |
|
|
|
37,440 |
|
The decrease in assets at December 31, 2016 compared with December 31, 2015 due to a decrease in cash and cash equivalents as these assets have been used to fund operations and a decrease in prepaid assets related to the drawdown of our escrow payments to our clinical research organization vendors. The decrease in liabilities at December 31, 2016 compared with December 31, 2015 was due to a decrease in clinical trial accruals associated with patient treatment costs in the AFFINITY trial, ENSPIRIT trial and our investigator sponsored trials evaluating apatorsen, lower deferred revenue as these amounts were recognized into collaboration revenue on a dollar for dollar basis as costs were incurred as part of the continuing research and development activities related to custirsen, the reversal of the lease termination liability and decrease in accrued compensation liabilities. This was partially offset by higher accrued liabilities as a result of the severance associated with the restructurings announced in fiscal 2016.
Critical Accounting Policies and Estimates
Use of Estimates
The preparation of consolidated financial statements in conformity with United States generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and notes thereto. Actual results could differ from these estimates. Estimates and assumptions principally relate to estimates of the fair value of our warrant liability, the initial fair value and forfeiture rates of stock options issued to employees and consultants, the estimated compensation cost on performance restricted stock unit awards and clinical trial and manufacturing accruals, estimated useful lives of property, plant and equipment and estimates and assumptions in contingent liabilities.
Fair value of financial instruments
The fair value of our cash equivalents and marketable securities is based on quoted market prices and trade data for comparable securities. We determine the fair value of our warrant liability based on the Black-Scholes pricing model and using considerable judgment, including estimating stock price volatility and expected warrant life. Other financial instruments including amounts receivable, accounts payable, accrued liabilities other, accrued clinical liabilities, accrued compensation and lease termination liability are carried at cost, which we believe approximates fair value because of the short-term maturities of these instruments.
Revenue Recognition
Revenue recognized to date is attributable to the upfront payment we received in the fourth quarter of 2009 pursuant to the collaboration agreement with Teva, as well as cash reimbursements from Teva for costs incurred by us under the clinical development plan. In April 2015, we and Teva entered into an agreement, or the Termination Agreement, pursuant to which the Collaboration Agreement was terminated and we regained rights to custirsen.
Pursuant to the Termination Agreement, Teva paid to us, as advanced reimbursement for certain continuing research and development activities related to custirsen, an amount equal to $27.0 million less approximately $3.8 million, which reduction represented a hold-back amount of $3.0 million and $0.8 million for certain third-party expenses incurred by Teva between January 1, 2015 and April 24, 2015, or Closing Date. Teva was permitted to deduct from the $3.0 million hold-back certain costs incurred after January 1, 2015 that arose after the Closing Date. Teva is responsible for expenses related to custirsen incurred pursuant to the Collaboration Agreement through December 31, 2014. We are responsible for certain custirsen-relate d expenses from and after January 1, 2015. Pursuant to the Termination Agreement, we received a nominal amount from the remaining hold-back after deductions by Teva for certain costs incurred after the Closing Date. We do not expect to receive any addition al amounts from Teva.
As a result of the termination of the Collaboration Agreement with Teva, we do not expect to earn any additional collaboration revenue beyond the amounts provided as advanced reimbursement for custirsen -related development expenses as set forth in the Termination Agreement. The advanced reimbursement payment made by Teva, as part of the Termination Agreement, was deferred and was recognized as collaboration revenue on a dollar for dollar basis as costs were incurred as part the of continuing research and development activities related to custirsen and certain other antisense inhibitors of clusterin. We have fully utilized the $23.2 million in advance reimbursement for custirsen-related development costs between January 1, 2015 and June 30, 2016.
47
Prior to the termination of the collaboration agreement, we and Teva shared certain custirsen-related development costs. We had spent the required $30 million in direct and indirect development costs, such as full-time equivalent (FTE) reimbursement for time incurred by our personnel for the benefit of the custirsen development plan. Teva funded all other expenses under the collaboration agreement including the three phase 3 clinical trials under the clinical development plan. On a quarterly basis Teva reimbursed all development expenses incurred in accordance with our clinical development plan. Our policy was to account for these reimbursements as Collaboration Revenue. For a summary description of the collaboration agreement with Teva see also Note 4.
The terminated collaboration agreement contained multiple elements and deliverables, and required evaluation pursuant to ASC 605-25, Multiple-Element Arrangements, or ASC 605-25. We evaluated the facts and circumstances of the collaboration agreement to determine whether we had obligations constituting deliverables under ASC 605-25. We concluded that we had multiple deliverables under the collaboration agreement, including deliverables relating to the grant of a technology license, and performance of manufacturing, regulatory and clinical development services in the U.S. and Canada, and estimated that the period in which it would perform those deliverables began in the fourth quarter of 2009 and was completed in the fourth quarter of 2012. Because we have been able to establish vendor specific objective evidence, or VSOE, of the fair value of the maintenance, regulatory, and clinical services, we concluded that these deliverables should be accounted as separate units of accounting under ASC 605-25. In establishing VSOE for the manufacturing, regulatory, and clinical development services, management relied on rates charged by other service providers providing similar development services.
Because we were not able to reliably estimate the fair value of the technology license, we used the residual value approach to determine the amount of revenue to recognize. Based on this approach, we recognized $22 million in 2009 relating to this element.
Impairment of Long-Lived Assets
We review long-lived assets for impairment whenever events or changes in circumstances indicate that the asset’s carrying amount may not be recoverable. We conduct our long-lived asset impairment analyses in accordance with ASC 360-10-15, “Impairment or Disposal of Long-Lived Assets.” ASC 360-10-15 requires us to group assets and liabilities at the lowest level for which identifiable cash flows are largely independent of the cash flows of other assets and liabilities and evaluate the asset group against the sum of the undiscounted future cash flows. If the undiscounted cash flows do not indicate the carrying amount of the asset is recoverable, an impairment charge is measured as the amount by which the carrying amount of the asset group exceeds its fair value based on discounted cash flow analysis or appraisals.
Income Taxes
Income taxes are accounted for under the liability method. Deferred tax assets and liabilities are recognized for the differences between the carrying values of assets and liabilities and their respective income tax bases and for operating losses and tax credit carry forwards. A valuation allowance is provided for the portion of deferred tax assets that is more likely than not to be unrealized. Deferred tax assets and liabilities are measured using the enacted tax rates and laws.
Scientific Research and Development Tax Credits
The benefits of tax credits for scientific research and development expenditures are recognized in the year the qualifying expenditure is made provided there is reasonable assurance of recoverability. The tax credits recorded are based on our estimates of amounts expected to be recovered and are subject to audit by taxation authorities. The non-refundable tax credit reduces the tax provision; however, no reduction to the tax provision has been recorded to date as we record a full valuation allowance. All qualifying expenditures are eligible for non-refundable tax credits only.
Research and Development Costs
Research and development costs are expensed as incurred, net of related refundable investment tax credits, with the exception of non-refundable advanced payments for goods or services to be used in future research and development, which are capitalized in accordance with ASC 730, “Research and Development” and included within Prepaid Expenses or Other Assets depending on when the assets will be utilized.
Clinical trial expenses are a component of research and development costs. These expenses include fees paid to contract research organizations and investigators and other service providers, which conduct certain product development activities on our behalf. We use an accrual basis of accounting, based upon estimates of the amount of service completed. In the event payments differ from the amount of service completed, prepaid expense or accrued liabilities amounts are adjusted on the balance sheet. These expenses are
48
based on estimates of the work performed under service agreements, milestones achieved, patient enrollment and experience with similar contracts. We monitor each of these factors to the extent possible and adjusts estimates accordingly.
Stock-Based Compensation
Effective January 1, 2006, we adopted the fair value recognition provisions of the ASC 718, “Stock Compensation”, using the modified prospective method with respect to options granted to employees and directors. Under this transition method, compensation cost is recognized in the financial statements beginning with the effective date for all share-based payments granted after January 1, 2006 and for all awards granted prior to but not yet vested as of January 1, 2006. The expense is amortized on a straight-line basis over the graded vesting period.
Restricted Stock Unit Awards
We grant restricted stock unit awards that generally vest and are expensed over a four-year period. We also granted restricted stock unit awards that vest in conjunction with certain performance conditions to certain executive officers and key employees. At each reporting date, we evaluate whether achievement of the performance conditions is probable. Compensation expense is recorded over the appropriate service period based upon our assessment of accomplishing each performance provision or the occurrence of other events that may have caused the awards to accelerate and vest.
Segment Information
We follow the requirements of ASC 280, “Segment Reporting.” We have one operating segment, dedicated to the development and commercialization of new cancer therapies, with operations located in Canada and the United States.
Warrants
We account for warrants pursuant to the authoritative guidance on accounting for derivative financial instruments indexed to, and potentially settled in, a company’s own stock, on the understanding that in compliance with applicable securities laws, the warrants require the issuance of registered securities upon exercise and therefore do not sufficiently preclude an implied right to net cash settlement. We classify warrants on the consolidated balance sheet as a liability which is revalued at each balance sheet date subsequent to the initial issuance. We also have warrants classified as equity and these are not reassessed for their fair value at the end of each reporting period. Warrants classified as equity are initially measured at their fair value and recognized as part of stockholders’ equity. Determining the appropriate fair-value model and calculating the fair value of registered warrants requires considerable judgment, including estimating stock price volatility and expected warrant life. The computation of expected volatility was based on the historical volatility of shares of our common stock for a period that coincides with the expected life of the warrants. A small change in the estimates used may have a relatively large change in the estimated valuation. We use the Black-Scholes pricing model to value the warrants. Changes in the fair value of the warrants classified as liabilities are reflected in the consolidated statement of loss as gain (loss) on revaluation of warrants.
Foreign Currency Translation
Our functional and reporting currency is the U.S. dollar. Revenues and expenses denominated in other than U.S. dollars are translated at average monthly rates.
The functional currency of our foreign subsidiary is the U.S. dollar. For this foreign operation, assets and liabilities denominated in other than U.S. dollars are translated at the period-end rates for monetary assets and liabilities and historical rates for non-monetary assets and liabilities. Revenues and expenses denominated in other than U.S. dollars are translated at average monthly rates. Gains and losses from this translation are recognized in the consolidated statement of loss.
Pending Adoption of Recent Accounting Pronouncements
On February 2016, the Financial Accounting Standards Board ("FASB") issued its new leases standard, ASU No. 2016-02, Leases (Topic 842) ("ASU 2016-02"). ASU 2016-02 is aimed at putting most leases on lessees’ balance sheets, but it would also change aspects of lessor accounting. ASU 2016-02 is effective for public business entities for annual periods beginning after December 15, 2018 and interim periods within that year. This standard is expected to have a significant impact on our current accounting for our lease arrangements, particularly our current operating lease arrangements, as well as, disclosures. We are currently evaluating the impact of adoption on our financial position and results from operations.
49
In May 2014, the FASB, issued ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606): Revenue from Contracts with Customers, which guidance in this update will supersede the revenue recognition requirements in Topic 605, Revenue Recognition, and most industry-specific guidance when it becomes effective. ASU No. 2014-09 affects any entity that enters into contracts with customers to transfer goods or services or enters into contracts for the transfer of nonfinancial assets unless those contracts are within the scope of other standards. The core principal of ASU No. 2014-09 is that a company will recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. In doing so, companies will need to use more judgment and make more estimates than under current guidance. These may include identifying performance obligations in the contract, estimating the amount of variable consideration to include in the transaction price and allocating the transaction price to each separate performance obligation. ASU No. 2014-09 is effective for annual reporting periods beginning after December 15, 2017, including interim periods within that reporting period, which will be our fiscal year 2018 (or December 31, 2018), and entities can transition to the standard either retrospectively or as a cumulative-effect adjustment as of the date of adoption. Early adoption is permitted. We are currently in the process of evaluating the impact of adoption of ASU No. 2014-09 and cannot reasonably estimate how the adoption of the standard will impact our consolidated financial statements and related disclosures.
Recently Adopted Accounting Policies
In March 2016, the FASB issued ASU 2016-09, Improvements to Employee Share-Based Payment Accounting. ASU 2016-09 simplifies several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. Some of the areas for simplification apply only to nonpublic entities. For public business entities, the amendments in this Update are effective for annual periods beginning after 15 December 2016, and interim periods within those annual periods. For all other entities, the amendments are effective for annual periods beginning after 15 December 2017, and interim periods within annual periods beginning after 15 December 2018. The adoption of this standard did not have a significant impact on our financial position or results of operations.
In November 2015, the FASB issued ASU No. 2015-17, Income Taxes (Topic 740): Balance Sheet Classification of Deferred Taxes. The standard requires that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. Entities are currently required to separate deferred income tax liabilities and assets into current and noncurrent amounts in a classified statement of financial position. The amendments, which require non-current presentation only (by jurisdiction), are effective for financial statements issued for annual periods beginning after December 15, 2016 with earlier application permitted as of the beginning of an interim or annual reporting period. The guidance is to be applied either prospectively to all deferred tax liabilities and assets or retrospectively to all periods presented. The adoption of this standard did not have a significant impact on our financial position or results of operations.
In February 2015, the FASB issued ASU 2015-02, Consolidation (Topic 810) — Amendments to the Consolidation Analysis. ASU 2015-02 eliminates the deferral of FAS 167 and makes changes to both the variable interest model and the voting model. For public business entities, the guidance is effective for annual and interim periods beginning after 15 December 2015. For nonpublic business entities, it is effective for annual periods beginning after 15 December 2016, and interim periods beginning after 15 December 2017. The adoption of this standard did not have a significant impact on our financial position or results of operations.
In January 2015, the FASB issued ASU 2015-01, Income Statement—Extraordinary and Unusual Items (Subtopic 225-20): Simplifying Income Statement Presentation by Eliminating the Concept of Extraordinary Items. ASU 2015-01 eliminates the concept of reporting extraordinary items, but retains current presentation and disclosure requirements for an event or transaction that is of an unusual nature or of a type that indicates infrequency of occurrence. Transactions that meet both criteria would now also follow such presentation and disclosure requirements. For all entities, the guidance is effective for annual periods, and interim periods within those annual periods, beginning after 15 December 2015. The adoption of this standard did not have a significant impact on our financial position or results of operations.
In August 2014, the Financial Accounting Standards Board, or FASB issued Accounting Standards Updated, or ASU No. 2014-15, Presentation of Financial Statements-Going Concern (Subtopic 2015-40) (ASU 2014-15). ASU 2014-15 provides guidance to U.S. GAAP about management’s responsibility to evaluate whether there is a substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. This new rule requires management to assess an entity’s ability to continue as a going concern by incorporating and expanding upon certain principles currently in the U.S. auditing standards. Specifically, ASU 2014-15 (1) defines the term substantial doubt, (2) requires an evaluation of every reporting period including interim periods, (3) provides principles for considering the mitigating effect of management’s plans, (5) requires an express statement and other disclosures when substantial doubt is not alleviated, and (6) requires an assessment for a period of one year after the date that the financial statements are issued (or available to be issued). This guidance is effective for annual periods ending after December 15, 2016. The adoption of this standard did not have a significant impact on our financial statement disclosures.
50
Interest Rate Risk
Interest rate risk is the risk that the fair values and future cash flows of financial instruments will fluctuate because of the changes in market interest rates. We invest our cash in a variety of financial instruments, primarily in short-term bank deposits, money market funds, and domestic and foreign commercial paper and government securities. These investments are denominated in U.S. dollars, and we monitor our exposure to interest rate changes is monitored. We have very limited interest rate risk due to the few assets or liabilities subject to fluctuations in interest rates. Our investment portfolio includes only marketable securities with active secondary or resale markets to help ensure portfolio liquidity. Due to the nature of our highly liquid marketable securities, a change in interest rates would not materially change the fair market value. We have estimated the effect on our portfolio of a hypothetical increase in interest rates by one percent to be a reduction of $0.2 million in the fair value of our investments as of December 31, 2016.
Foreign Currency Exchange Risk
We are exposed to risks associated with foreign currency transactions on certain contracts and payroll expenses related to our Canadian subsidiary, OncoGenex Technologies, denominated in Canadian dollars and we have not hedged these amounts. As our unhedged foreign currency transactions fluctuate, our earnings might be negatively affected. Accordingly, changes in the value of the U.S. dollar relative to the Canadian dollar might have an adverse effect on our reported results of operations and financial condition, and fluctuations in exchange rates might harm our reported results and accounts from period to period. We have estimated the effect on our reported results of operations of a hypothetical increase of 10 percent in the exchange rate of the Canadian dollar against the U.S. dollar to be $0.3 million for the year ended December 31, 2016.
51
INDEX TO FINANCIAL STATEMENTS:
52
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of
OncoGenex Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of OncoGenex Pharmaceuticals, Inc. (the “Company”) as of December 31, 2016 and 2015, and the related consolidated statements of loss and comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2016. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of OncoGenex Pharmaceuticals, Inc. at December 31, 2016 and 2015, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2016, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), OncoGenex Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2016, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework) and our report dated February 23, 2017 expressed an unqualified opinion thereon.
|
Vancouver, Canada |
|
|
/s/ ERNST & YOUNG LLP |
|
|
February 23, 2017 |
|
|
Chartered Professional Accountants |
|
53
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of
OncoGenex Pharmaceuticals, Inc.
We have audited OncoGenex Pharmaceuticals, Inc.’s (the “Company”) internal control over financial reporting as of December 31, 2016, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework) (the COSO criteria). OncoGenex Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying “Management’s Report on Internal Control over Financial Reporting”. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, OncoGenex Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2016, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of OncoGenex Pharmaceuticals, Inc. as of December 31, 2016 and 2015, and the related consolidated statements of loss and comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2016 and our report dated February 23, 2017 expressed an unqualified opinion thereon.
|
Vancouver, Canada |
|
|
/s/ ERNST & YOUNG LLP |
|
|
February 23, 2017 |
|
|
Chartered Professional Accountants |
|
54
OncoGenex Pharmaceuticals, Inc.
(In thousands, except per share and share amounts)
|
|
|
December 31, |
|
|||||
|
|
|
2016 |
|
|
2015 |
|
||
|
ASSETS |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents [note 5] |
|
$ |
15,233 |
|
|
$ |
34,310 |
|
|
Short-term investments [note 5] |
|
|
10,230 |
|
|
|
20,876 |
|
|
Interest receivable |
|
|
32 |
|
|
|
111 |
|
|
Amounts receivable |
|
|
478 |
|
|
|
14 |
|
|
Prepaid expenses |
|
|
954 |
|
|
|
1,987 |
|
|
Total current assets |
|
|
26,927 |
|
|
|
57,298 |
|
|
Restricted cash [note 5] |
|
|
272 |
|
|
|
272 |
|
|
Property and equipment, net [note 6] |
|
|
258 |
|
|
|
602 |
|
|
Other assets [note 8] |
|
|
13 |
|
|
|
37 |
|
|
Total assets |
|
$ |
27,470 |
|
|
$ |
58,209 |
|
|
LIABILITIES AND STOCKHOLDERS' EQUITY |
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
2,121 |
|
|
$ |
1,343 |
|
|
Accrued liabilities other |
|
|
2,442 |
|
|
|
641 |
|
|
Accrued clinical liabilities |
|
|
3,415 |
|
|
|
9,966 |
|
|
Accrued compensation |
|
|
188 |
|
|
|
1,267 |
|
|
Current portion of long-term obligations [note 12] |
|
|
57 |
|
|
|
52 |
|
|
Lease termination liability [note 7] |
|
|
— |
|
|
|
1,250 |
|
|
Deferred collaboration revenue [note 4] |
|
|
— |
|
|
|
5,040 |
|
|
Warrant liability [note 5 and note 10] |
|
|
232 |
|
|
|
1,105 |
|
|
Total current liabilities |
|
|
8,455 |
|
|
|
20,664 |
|
|
Long-term obligations, less current portion [note 12] |
|
|
49 |
|
|
|
105 |
|
|
Total liabilities |
|
|
8,504 |
|
|
|
20,769 |
|
|
Commitments and contingencies [note 4 and note 12] |
|
|
|
|
|
|
|
|
|
Stockholders' equity: |
|
|
|
|
|
|
|
|
|
Common stock, $0.001 par value, 75,000,000 shares authorized, 30,059,514 and 29,846,991 issued at December 31, 2016 and December 31, 2015, respectively, and 30,025,521 and 29,812,998 outstanding at December 31, 2016 and December 31, 2015, respectively |
|
|
29 |
|
|
|
29 |
|
|
Additional paid-in capital |
|
|
213,239 |
|
|
|
211,590 |
|
|
Accumulated deficit |
|
|
(196,942 |
) |
|
|
(176,811 |
) |
|
Accumulated other comprehensive income |
|
|
2,640 |
|
|
|
2,632 |
|
|
Total stockholders' equity |
|
|
18,966 |
|
|
|
37,440 |
|
|
Total liabilities and stockholders' equity |
|
$ |
27,470 |
|
|
$ |
58,209 |
|
|
Subsequent events [note 15] |
|
|
|
|
|
|
|
|
See accompanying notes.
55
OncoGenex Pharmaceuticals, Inc.
Consolidated Statements of Loss and Comprehensive Loss
(In thousands, except per share and share amounts)
|
|
|
Year Ended |
|
|||||||||
|
|
|
December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
COLLABORATION REVENUE [note 4] |
|
$ |
5,062 |
|
|
$ |
18,160 |
|
|
$ |
27,116 |
|
|
EXPENSES |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
14,788 |
|
|
|
25,108 |
|
|
|
46,224 |
|
|
General and administrative |
|
|
8,933 |
|
|
|
11,805 |
|
|
|
10,625 |
|
|
Restructuring costs (recovery) [note 13] |
|
|
2,206 |
|
|
|
— |
|
|
|
(267 |
) |
|
Recovery of lease termination loss [note 12] |
|
|
(1,250 |
) |
|
|
— |
|
|
|
— |
|
|
Litigation settlement [note 12] |
|
|
1,375 |
|
|
|
— |
|
|
|
— |
|
|
Asset impairment charge [note 6] |
|
|
202 |
|
|
|
— |
|
|
|
— |
|
|
Total operating expenses |
|
|
26,254 |
|
|
|
36,913 |
|
|
|
56,582 |
|
|
OTHER INCOME (EXPENSE) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
203 |
|
|
|
119 |
|
|
|
35 |
|
|
Other income (expense) |
|
|
(13 |
) |
|
|
(64 |
) |
|
|
(19 |
) |
|
Warrant issuance costs |
|
|
— |
|
|
|
— |
|
|
|
(531 |
) |
|
Gain on warrants [note 5 and note 10[e]] |
|
|
873 |
|
|
|
1,897 |
|
|
|
3,741 |
|
|
Total other income |
|
|
1,063 |
|
|
|
1,952 |
|
|
|
3,226 |
|
|
Net loss |
|
$ |
(20,129 |
) |
|
$ |
(16,801 |
) |
|
$ |
(26,240 |
) |
|
OTHER COMPREHENSIVE INCOME |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net unrealized gain (loss) on securities |
|
|
8 |
|
|
|
10 |
|
|
|
(21 |
) |
|
Total other comprehensive income (loss) |
|
|
8 |
|
|
|
10 |
|
|
|
(21 |
) |
|
Comprehensive loss |
|
$ |
(20,121 |
) |
|
$ |
(16,791 |
) |
|
$ |
(26,261 |
) |
|
Basic and diluted net loss per common share [note 10 [g]] |
|
$ |
(0.67 |
) |
|
$ |
(0.64 |
) |
|
$ |
(1.45 |
) |
|
Shares used in computation of basic and diluted net loss per common share [note 10 [g]] |
|
|
29,949,432 |
|
|
|
26,147,344 |
|
|
|
18,098,799 |
|
See accompanying notes.
56
OncoGenex Pharmaceuticals, Inc.
Consolidated Statements of Stockholders’ Equity
(In thousands, except share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional |
|
|
Other |
|
|
|
|
|
|
Total, |
|
|||
|
|
|
Common Stock |
|
|
Paid-in |
|
|
Comprehensive |
|
|
Accumulated |
|
|
Stockholders |
|
|||||||||
|
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Income (Loss) |
|
|
Deficit |
|
|
Equity |
|
||||||
|
Balance, January 1, 2014 |
|
|
14,707,886 |
|
|
$ |
15 |
|
|
$ |
168,242 |
|
|
$ |
2,643 |
|
|
$ |
(133,689 |
) |
|
|
37,211 |
|
|
Stock-based compensation expense |
|
|
— |
|
|
|
— |
|
|
|
3,860 |
|
|
|
— |
|
|
|
— |
|
|
|
3,860 |
|
|
Stock option exercises |
|
|
10,000 |
|
|
|
— |
|
|
|
30 |
|
|
|
— |
|
|
|
— |
|
|
|
30 |
|
|
Restricted Stock Unit Settlements |
|
|
53,971 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Performance Restricted Stock Unit Settlements |
|
|
149,177 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Performance Restricted Stock Unit Settlements withheld and retired to treasury |
|
|
(9,226 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(29 |
) |
|
|
(29 |
) |
|
Shares issued - ATM Financing |
|
|
809,214 |
|
|
|
— |
|
|
|
2,860 |
|
|
|
— |
|
|
|
— |
|
|
|
2,860 |
|
|
Shares issued in July 2014 Financing |
|
|
5,559,866 |
|
|
|
6 |
|
|
|
16,369 |
|
|
|
— |
|
|
|
— |
|
|
|
16,375 |
|
|
Warrant Exercises |
|
|
1,340,538 |
|
|
|
1 |
|
|
|
12 |
|
|
|
— |
|
|
|
— |
|
|
|
13 |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(26,240 |
) |
|
|
(26,240 |
) |
|
Other comprehensive income (loss) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(21 |
) |
|
|
— |
|
|
|
(21 |
) |
|
Balance, December 31, 2014 |
|
|
22,621,426 |
|
|
|
22 |
|
|
|
191,373 |
|
|
|
2,622 |
|
|
|
(159,958 |
) |
|
|
34,059 |
|
|
Stock-based compensation expense |
|
|
— |
|
|
|
— |
|
|
|
2,328 |
|
|
|
— |
|
|
|
— |
|
|
|
2,328 |
|
|
Stock option exercises |
|
|
5,359 |
|
|
|
— |
|
|
|
14 |
|
|
|
— |
|
|
|
— |
|
|
|
14 |
|
|
Restricted Stock Unit Settlements |
|
|
186,991 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Performance Restricted Stock Unit Settlements |
|
|
82,410 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Performance Restricted Stock Unit Settlements withheld and retired to treasury |
|
|
(24,750 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(52 |
) |
|
|
(52 |
) |
|
Shares issues - Lincoln Park Capital |
|
|
6,941,562 |
|
|
|
7 |
|
|
|
17,875 |
|
|
|
— |
|
|
|
— |
|
|
|
17,882 |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(16,801 |
) |
|
|
(16,801 |
) |
|
Other comprehensive income (loss) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
10 |
|
|
|
— |
|
|
|
10 |
|
|
Balance, December 31, 2015 |
|
|
29,812,998 |
|
|
|
29 |
|
|
|
211,590 |
|
|
|
2,632 |
|
|
|
(176,811 |
) |
|
|
37,440 |
|
|
Stock-based compensation expense |
|
|
— |
|
|
|
— |
|
|
|
1,649 |
|
|
|
— |
|
|
|
— |
|
|
|
1,649 |
|
|
Restricted Stock Unit Settlements |
|
|
212,523 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(2 |
) |
|
|
(2 |
) |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(20,129 |
) |
|
|
(20,129 |
) |
|
Other comprehensive income (loss) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
8 |
|
|
|
— |
|
|
|
8 |
|
|
Balance, December 31, 2016 |
|
|
30,025,521 |
|
|
$ |
29 |
|
|
$ |
213,239 |
|
|
$ |
2,640 |
|
|
$ |
(196,942 |
) |
|
$ |
18,966 |
|
See accompanying notes.
57
OncoGenex Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(In thousands)
|
|
|
Year Ended |
|
|||||||||
|
|
|
December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Operating Activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(20,129 |
) |
|
$ |
(16,801 |
) |
|
$ |
(26,240 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Gain on warrants [note 5 and note 10[e]] |
|
|
(873 |
) |
|
|
(1,897 |
) |
|
|
(3,741 |
) |
|
Warrant issuance costs |
|
|
— |
|
|
|
— |
|
|
|
531 |
|
|
Depreciation |
|
|
188 |
|
|
|
244 |
|
|
|
223 |
|
|
Asset impairment charge [note 6] |
|
|
202 |
|
|
|
— |
|
|
|
— |
|
|
Stock-based compensation [note10[c]] |
|
|
1,649 |
|
|
|
2,328 |
|
|
|
3,860 |
|
|
Restructuring gain [note 7] |
|
|
— |
|
|
|
— |
|
|
|
(3,517 |
) |
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest receivable |
|
|
79 |
|
|
|
2 |
|
|
|
105 |
|
|
Amounts receivable |
|
|
(464 |
) |
|
|
5,662 |
|
|
|
2,981 |
|
|
Prepaid expenses and other assets |
|
|
1,057 |
|
|
|
1,167 |
|
|
|
3,944 |
|
|
Accounts payable |
|
|
778 |
|
|
|
1,271 |
|
|
|
(67 |
) |
|
Accrued liabilities other |
|
|
1,801 |
|
|
|
(222 |
) |
|
|
363 |
|
|
Accrued clinical liabilities |
|
|
(6,551 |
) |
|
|
(3,714 |
) |
|
|
2,178 |
|
|
Accrued compensation |
|
|
(1,079 |
) |
|
|
(66 |
) |
|
|
(372 |
) |
|
Restricted cash |
|
|
— |
|
|
|
(21 |
) |
|
|
63 |
|
|
Excess lease liability [note 7] |
|
|
— |
|
|
|
(194 |
) |
|
|
(785 |
) |
|
Lease obligation |
|
|
(51 |
) |
|
|
100 |
|
|
|
(84 |
) |
|
Lease termination fees [note 12] |
|
|
(1,250 |
) |
|
|
(2,000 |
) |
|
|
3,250 |
|
|
Deferred collaboration revenue [note 4] |
|
|
(5,040 |
) |
|
|
5,040 |
|
|
|
— |
|
|
Net cash used in operating activities |
|
|
(29,683 |
) |
|
|
(9,101 |
) |
|
|
(17,308 |
) |
|
Financing Activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from the exercise of stock options |
|
|
— |
|
|
|
14 |
|
|
|
30 |
|
|
Proceeds from ATM Financing, net of issuance costs |
|
|
— |
|
|
|
17,629 |
|
|
|
2,874 |
|
|
Taxes paid related to net share settlement of equity awards |
|
|
(2 |
) |
|
|
(52 |
) |
|
|
(29 |
) |
|
Issuance of common shares, net of share issuance costs |
|
|
— |
|
|
|
— |
|
|
|
22,372 |
|
|
Net cash provided by (used in) financing activities |
|
|
(2 |
) |
|
|
17,591 |
|
|
|
25,247 |
|
|
Investing Activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of investments |
|
|
(36,504 |
) |
|
|
(24,368 |
) |
|
|
(19,446 |
) |
|
Proceeds from sale of investments |
|
|
— |
|
|
|
1,003 |
|
|
|
— |
|
|
Proceeds from maturities of investments |
|
|
47,158 |
|
|
|
21,659 |
|
|
|
24,894 |
|
|
Purchase of property and equipment |
|
|
(46 |
) |
|
|
(371 |
) |
|
|
(82 |
) |
|
Net cash provided by (used in) investing activities |
|
|
10,608 |
|
|
|
(2,077 |
) |
|
|
5,366 |
|
|
Effect of exchange rate changes on cash |
|
|
— |
|
|
|
— |
|
|
|
(1 |
) |
|
Net increase (decrease) in cash and cash equivalents |
|
|
(19,077 |
) |
|
|
6,413 |
|
|
|
13,304 |
|
|
Cash and cash equivalents at beginning of year |
|
|
34,310 |
|
|
|
27,897 |
|
|
|
14,593 |
|
|
Cash and cash equivalents at end of year |
|
$ |
15,233 |
|
|
$ |
34,310 |
|
|
$ |
27,897 |
|
|
Supplemental Disclosure of Cash Flow Information: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment received and accrued but not yet paid |
|
$ |
— |
|
|
$ |
218 |
|
|
$ |
— |
|
See accompanying notes.
58
OncoGenex Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
(In thousands, except per share and share amounts)
1. NATURE OF BUSINESS AND BASIS OF PRESENTATION
OncoGenex Pharmaceuticals, Inc. (referred to as “OncoGenex,” “we,” “us,” or “our”) is committed to the development and commercialization of new therapies that address treatment resistance in cancer patients. We were incorporated in the state of Delaware and are headquartered in Bothell, Washington and have a subsidiary in Vancouver, British Columbia.
Basis of Presentation
The consolidated financial statements include the accounts of OncoGenex and our wholly owned subsidiary, OncoGenex Technologies Inc., or OncoGenex Technologies. All intercompany balances and transactions have been eliminated.
Liquidity
We have historically experienced recurring losses from operations that have generated an accumulated deficit of $196.9 million through December 31, 2016. At December 31, 2016, we had cash, cash equivalents and short-term investments of $25.5 million.
2. ACCOUNTING POLICIES
Significant Accounting Policies
Use of Estimates
The preparation of consolidated financial statements in conformity with United States generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and notes thereto. Actual results could differ from these estimates. Estimates and assumptions principally relate to estimates of the fair value of our warrant liability, the initial fair value and forfeiture rates of stock options issued to employees and consultants, the estimated compensation cost on performance restricted stock unit awards and clinical trial and manufacturing accruals, estimated useful lives of property, plant and equipment and estimates and assumptions in contingent liabilities.
Cash Equivalents
We consider all highly liquid investments with an original maturity of three months or less to be cash equivalents, which we consider as available for sale and carry at fair value, with unrealized gains and losses, if any, reported as accumulated other comprehensive income or loss, which is a separate component of stockholders’ equity.
Short-Term Investments
Short-term investments consist of financial instruments purchased with an original maturity of greater than three months and less than one year. We consider our short-term investments as available-for-sale and carry them at fair value, with unrealized gains and losses except other than temporary losses, if any, reported as accumulated other comprehensive income or loss, which is a separate component of stockholders’ equity. Realized gains and losses on the sale of these securities are recognized in net income or loss. The cost of investments sold is based on the specific identification method.
Fair value of financial instruments
The fair value of our cash equivalents and marketable securities is based on quoted market prices and trade data for comparable securities. We determine the fair value of our warrant liability based on the Black-Scholes pricing model and using considerable judgment, including estimating stock price volatility and expected warrant life. Other financial instruments including amounts receivable, accounts payable, accrued liabilities other, accrued clinical liabilities, accrued compensation and lease termination liability are carried at cost, which we believe approximates fair value because of the short-term maturities of these instruments.
59
The costs of acquiring intellectual property rights to be used in the research and development process, including licensing fees and milestone payments, are charged to research and development expense as incurred in situations where we have not identified an alternative future use for the acquired rights, and are capitalized in situations where we have identified an alternative future use. No costs associated with acquiring intellectual property rights have been capitalized to date. Costs of maintaining intellectual property rights are expensed as incurred.
Revenue Recognition
Revenue recognized to date is attributable to the upfront payment we received in the fourth quarter of 2009 pursuant to the collaboration agreement with Teva, as well as cash reimbursements from Teva for costs incurred by us under the clinical development plan. In April 2015, we and Teva entered into an agreement, or the Termination Agreement, pursuant to which the Collaboration Agreement was terminated and we regained rights to custirsen.
Pursuant to the Termination Agreement, Teva paid to us, as advanced reimbursement for certain continuing research and development activities related to custirsen, an amount equal to $27.0 million less approximately $3.8 million, which reduction represented a hold-back amount of $3.0 million and $0.8 million for certain third-party expenses incurred by Teva between January 1, 2015 and April 24, 2015, or Closing Date. Teva was permitted to deduct from the $3.0 million hold-back certain costs incurred after January 1, 2015 that arose after the Closing Date. Teva is responsible for expenses related to custirsen incurred pursuant to the Collaboration Agreement through December 31, 2014. We are responsible for certain custirsen-relate d expenses from and after January 1, 2015. Pursuant to the Termination Agreement, we received a nominal amount from the remaining hold-back after deductions by Teva for certain costs incurred after the Closing Date. We do not expect to receive any addition al amounts from Teva.
As a result of the termination of the Collaboration Agreement with Teva, we do not expect to earn any additional collaboration revenue beyond the amounts provided as advanced reimbursement for custirsen -related development expenses as set forth in the Termination Agreement. The advanced reimbursement payment made by Teva, as part of the Termination Agreement, was deferred and was recognized as collaboration revenue on a dollar for dollar basis as costs were incurred as part the of continuing research and development activities related to custirsen and certain other antisense inhibitors of clusterin. We have fully utilized the $23.2 million in advance reimbursement for custirsen-related development costs between January 1, 2015 and June 30, 2016.
Prior to the termination of the collaboration agreement, we and Teva shared certain custirsen-related development costs. We had spent the required $30 million in direct and indirect development costs, such as full-time equivalent (FTE) reimbursement for time incurred by our personnel for the benefit of the custirsen development plan. Teva funded all other expenses under the collaboration agreement including the three phase 3 clinical trials under the clinical development plan. On a quarterly basis Teva reimbursed all development expenses incurred in accordance with our clinical development plan. Our policy was to account for these reimbursements as Collaboration Revenue. For a summary description of the collaboration agreement with Teva see also Note 4.
The terminated collaboration agreement contained multiple elements and deliverables, and required evaluation pursuant to ASC 605-25, Multiple-Element Arrangements, or ASC 605-25. We evaluated the facts and circumstances of the collaboration agreement to determine whether we had obligations constituting deliverables under ASC 605-25. We concluded that we had multiple deliverables under the collaboration agreement, including deliverables relating to the grant of a technology license, and performance of manufacturing, regulatory and clinical development services in the U.S. and Canada, and estimated that the period in which it would perform those deliverables began in the fourth quarter of 2009 and was completed in the fourth quarter of 2012. Because we have been able to establish vendor specific objective evidence, or VSOE, of the fair value of the maintenance, regulatory, and clinical services, we concluded that these deliverables should be accounted as separate units of accounting under ASC 605-25. In establishing VSOE for the manufacturing, regulatory, and clinical development services, management relied on rates charged by other service providers providing similar development services.
Because we were not able to reliably estimate the fair value of the technology license, we used the residual value approach to determine the amount of revenue to recognize. Based on this approach, we recognized $22 million in 2009 relating to this element.
60
Property and equipment assets are recorded at cost less accumulated depreciation. Depreciation expense on assets acquired under capital lease is recorded within depreciation expense. Depreciation is recorded on a straight-line basis over the following periods:
|
Computer equipment |
|
3 years |
|
Furniture and fixtures |
|
5 years |
|
Machinery and equipment |
|
5 - 10 years |
|
Leasehold improvements and equipment under capital lease |
|
Over the term of the lease |
Impairment of Long-Lived Assets
We review long-lived assets for impairment whenever events or changes in circumstances indicate that the asset’s carrying amount may not be recoverable. We conduct our long-lived asset impairment analyses in accordance with ASC 360-10-15, “Impairment or Disposal of Long-Lived Assets.” ASC 360-10-15 requires us to group assets and liabilities at the lowest level for which identifiable cash flows are largely independent of the cash flows of other assets and liabilities and evaluate the asset group against the sum of the undiscounted future cash flows. If the undiscounted cash flows do not indicate the carrying amount of the asset is recoverable, an impairment charge is measured as the amount by which the carrying amount of the asset group exceeds its fair value based on discounted cash flow analysis or appraisals.
Income Taxes
Income taxes are accounted for under the liability method. Deferred tax assets and liabilities are recognized for the differences between the carrying values of assets and liabilities and their respective income tax bases and for operating losses and tax credit carry forwards. A valuation allowance is provided for the portion of deferred tax assets that is more likely than not to be unrealized. Deferred tax assets and liabilities are measured using the enacted tax rates and laws.
Scientific Research and Development Tax Credits
The benefits of tax credits for scientific research and development expenditures are recognized in the year the qualifying expenditure is made provided there is reasonable assurance of recoverability. The tax credits recorded are based on our estimates of amounts expected to be recovered and are subject to audit by taxation authorities. The non-refundable tax credit reduces the tax provision; however, no reduction to the tax provision has been recorded to date as we record a full valuation allowance. All qualifying expenditures are eligible for non-refundable tax credits only.
Research and Development Costs
Research and development costs are expensed as incurred, net of related refundable investment tax credits, with the exception of non-refundable advanced payments for goods or services to be used in future research and development, which are capitalized in accordance with ASC 730, “Research and Development” and included within Prepaid Expenses or Other Assets depending on when the assets will be utilized.
Clinical trial expenses are a component of research and development costs. These expenses include fees paid to contract research organizations and investigators and other service providers, which conduct certain product development activities on our behalf. We use an accrual basis of accounting, based upon estimates of the amount of service completed. In the event payments differ from the amount of service completed, prepaid expense or accrued liabilities amounts are adjusted on the balance sheet. These expenses are based on estimates of the work performed under service agreements, milestones achieved, patient enrollment and experience with similar contracts. We monitor each of these factors to the extent possible and adjusts estimates accordingly.
Stock-Based Compensation
Effective January 1, 2006, we adopted the fair value recognition provisions of the ASC 718, “Stock Compensation”, using the modified prospective method with respect to options granted to employees and directors. Under this transition method, compensation cost is recognized in the financial statements beginning with the effective date for all share-based payments granted after January 1, 2006 and for all awards granted prior to but not yet vested as of January 1, 2006. The expense is amortized on a straight-line basis over the graded vesting period.
61
We grant restricted stock unit awards that generally vest and are expensed over a four-year period. We also granted restricted stock unit awards that vest in conjunction with certain performance conditions to certain executive officers and key employees. At each reporting date, we evaluate whether achievement of the performance conditions is probable. Compensation expense is recorded over the appropriate service period based upon our assessment of accomplishing each performance provision or the occurrence of other events that may have caused the awards to accelerate and vest.
Segment Information
We follow the requirements of ASC 280, “Segment Reporting.” We have one operating segment, dedicated to the development and commercialization of new cancer therapies, with operations located in Canada and the United States.
Comprehensive Income (Loss)
Comprehensive income (loss) is comprised of net income (loss) and other comprehensive income (loss). Other comprehensive income (loss) consists of unrealized gains and losses on our available-for-sale marketable securities. We report the components of comprehensive loss in the statement of stockholders’ equity.
Loss per Common Share
Basic loss per common share is computed using the weighted average number of common shares outstanding during the period. Diluted loss per common share is computed in accordance with the treasury stock method. The effect of potentially issuable common shares from outstanding stock options, restricted stock unit awards and warrants are anti-dilutive for all periods presented.
Warrants
We account for warrants pursuant to the authoritative guidance on accounting for derivative financial instruments indexed to, and potentially settled in, a company’s own stock, on the understanding that in compliance with applicable securities laws, the warrants require the issuance of registered securities upon exercise and therefore do not sufficiently preclude an implied right to net cash settlement. We classify warrants on the consolidated balance sheet as a liability which is revalued at each balance sheet date subsequent to the initial issuance. We also have warrants classified as equity and these are not reassessed for their fair value at the end of each reporting period. Warrants classified as equity are initially measured at their fair value and recognized as part of stockholders’ equity. Determining the appropriate fair-value model and calculating the fair value of registered warrants requires considerable judgment, including estimating stock price volatility and expected warrant life. The computation of expected volatility was based on the historical volatility of shares of our common stock for a period that coincides with the expected life of the warrants. A small change in the estimates used may have a relatively large change in the estimated valuation. We use the Black-Scholes pricing model to value the warrants. Changes in the fair value of the warrants classified as liabilities are reflected in the consolidated statement of loss as gain (loss) on revaluation of warrants.
Foreign Currency Translation
Our functional and reporting currency is the U.S. dollar. Revenues and expenses denominated in other than U.S. dollars are translated at average monthly rates.
The functional currency of our foreign subsidiary is the U.S. dollar. For this foreign operation, assets and liabilities denominated in other than U.S. dollars are translated at the period-end rates for monetary assets and liabilities and historical rates for non-monetary assets and liabilities. Revenues and expenses denominated in other than U.S. dollars are translated at average monthly rates. Gains and losses from this translation are recognized in the consolidated statement of loss.
Pending Adoption of Recent Accounting Pronouncements
On February 2016, the Financial Accounting Standards Board ("FASB") issued its new leases standard, ASU No. 2016-02, Leases (Topic 842) ("ASU 2016-02"). ASU 2016-02 is aimed at putting most leases on lessees’ balance sheets, but it would also change aspects of lessor accounting. ASU 2016-02 is effective for public business entities for annual periods beginning after December 15, 2018 and interim periods within that year. This standard is expected to have a significant impact on our current accounting for our lease arrangements, particularly our current operating lease arrangements, as well as, disclosures. We are currently evaluating the impact of adoption on our financial position and results from operations.
62
In May 2014, the FASB, issued ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606): Revenue from Contracts with Customers, which guidance in this update will supersede the revenue recognition requirements in Topic 605, Revenue Recognition, and most industry-specific guidance when it becomes effective. ASU No. 2014-09 affects any entity that enters into contracts with customers to transfer goods or services or enters into contracts for the transfer of nonfinancial assets unless those contracts are within the scope of other standards. The core principal of ASU No. 2014-09 is that a company will recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the company expects to be entitled in exchange for those goods or services. In doing so, companies will need to use more judgment and make more estimates than under current guidance. These may include identifying performance obligations in the contract, estimating the amount of variable consideration to include in the transaction price and allocating the transaction price to each separate performance obligation. ASU No. 2014-09 is effective for annual reporting periods beginning after December 15, 2017, including interim periods within that reporting period, which will be our fiscal year 2018 (or December 31, 2018), and entities can transition to the standard either retrospectively or as a cumulative-effect adjustment as of the date of adoption. Early adoption is permitted. We are currently in the process of evaluating the impact of adoption of ASU No. 2014-09 and cannot reasonably estimate how the adoption of the standard will impact our consolidated financial statements and related disclosures.
Recently Adopted Accounting Policies
In March 2016, the FASB issued ASU 2016-09, Improvements to Employee Share-Based Payment Accounting. ASU 2016-09 simplifies several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. Some of the areas for simplification apply only to nonpublic entities. For public business entities, the amendments in this Update are effective for annual periods beginning after 15 December 2016, and interim periods within those annual periods. For all other entities, the amendments are effective for annual periods beginning after 15 December 2017, and interim periods within annual periods beginning after 15 December 2018. The adoption of this standard did not have a significant impact on our financial position or results of operations.
In November 2015, the FASB issued ASU No. 2015-17, Income Taxes (Topic 740): Balance Sheet Classification of Deferred Taxes. The standard requires that deferred tax liabilities and assets be classified as noncurrent in a classified statement of financial position. Entities are currently required to separate deferred income tax liabilities and assets into current and noncurrent amounts in a classified statement of financial position. The amendments, which require non-current presentation only (by jurisdiction), are effective for financial statements issued for annual periods beginning after December 15, 2016 with earlier application permitted as of the beginning of an interim or annual reporting period. The guidance is to be applied either prospectively to all deferred tax liabilities and assets or retrospectively to all periods presented. The adoption of this standard did not have a significant impact on our financial position or results of operations.
In February 2015, the FASB issued ASU 2015-02, Consolidation (Topic 810) — Amendments to the Consolidation Analysis. ASU 2015-02 eliminates the deferral of FAS 167 and makes changes to both the variable interest model and the voting model. For public business entities, the guidance is effective for annual and interim periods beginning after 15 December 2015. For nonpublic business entities, it is effective for annual periods beginning after 15 December 2016, and interim periods beginning after 15 December 2017. The adoption of this standard did not have a significant impact on our financial position or results of operations.
In January 2015, the FASB issued ASU 2015-01, Income Statement—Extraordinary and Unusual Items (Subtopic 225-20): Simplifying Income Statement Presentation by Eliminating the Concept of Extraordinary Items. ASU 2015-01 eliminates the concept of reporting extraordinary items, but retains current presentation and disclosure requirements for an event or transaction that is of an unusual nature or of a type that indicates infrequency of occurrence. Transactions that meet both criteria would now also follow such presentation and disclosure requirements. For all entities, the guidance is effective for annual periods, and interim periods within those annual periods, beginning after 15 December 2015. The adoption of this standard did not have a significant impact on our financial position or results of operations.
In August 2014, the Financial Accounting Standards Board, or FASB issued Accounting Standards Updated, or ASU No. 2014-15, Presentation of Financial Statements-Going Concern (Subtopic 2015-40) (ASU 2014-15). ASU 2014-15 provides guidance to U.S. GAAP about management’s responsibility to evaluate whether there is a substantial doubt about an entity’s ability to continue as a going concern and to provide related footnote disclosures. This new rule requires management to assess an entity’s ability to continue as a going concern by incorporating and expanding upon certain principles currently in the U.S. auditing standards. Specifically, ASU 2014-15 (1) defines the term substantial doubt, (2) requires an evaluation of every reporting period including interim periods, (3) provides principles for considering the mitigating effect of management’s plans, (5) requires an express statement and other disclosures when substantial doubt is not alleviated, and (6) requires an assessment for a period of one year after the date that the financial statements are issued (or available to be issued). This guidance is effective for annual periods ending after December 15, 2016. The adoption of this standard did not have a significant impact on our financial statement disclosures.
63
3. FINANCIAL INSTRUMENTS AND RISK
For certain of our financial instruments, including cash and cash equivalents, amounts receivable, accounts payable, accrued liabilities other, accrued clinical liabilities, accrued compensation and lease termination liability carrying values approximate fair value due to their short-term nature. Our cash equivalents and short-term investments are recorded at fair value.
Financial risk is the risk to our results of operations that arises from fluctuations in interest rates and foreign exchange rates and the degree of volatility of these rates as well as credit risk associated with the financial stability of the issuers of the financial instruments. Foreign exchange rate risk arises as a portion of our investments which finance operations and a portion of our expenses are denominated in other than U.S. dollars.
We invest our excess cash in accordance with investment guidelines, which limit our credit exposure to any one financial institution or corporation other than securities issued by the U.S. government. We only invest in A (or equivalent) rated securities with maturities of one year or less. These securities generally mature within one year or less and in some cases are not collateralized. At December 31, 2016, the average days to maturity of our portfolio of cash equivalents and marketable securities was 45 days (December 31, 2015 – 61 days). We do not use derivative instruments to hedge against any of these financial risks.
4. COLLABORATION AGREEMENT
In December 2009, we, through our wholly-owned subsidiary, OncoGenex Technologies, entered into a collaboration agreement, or Collaboration Agreement, with Teva Pharmaceutical Industries Ltd., or Teva, for the development and global commercialization of custirsen (and related compounds), a pharmaceutical compound designed to inhibit the production of clusterin, a protein we believe is associated with cancer treatment resistance, or the Licensed Product. In December 2014, we and Teva agreed to terminate the Collaboration Agreement upon entry into a termination agreement. In April 2015, we and Teva entered into an agreement, or the Termination Agreement, pursuant to which the Collaboration Agreement was terminated and we regained rights to custirsen.
Pursuant to the Termination Agreement, Teva paid to us, as advanced reimbursement for certain continuing research and development activities related to custirsen, an amount equal to $27.0 million less approximately $3.8 million, which reduction represented a hold-back amount of $3.0 million and $0.8 million for certain third-party expenses incurred by Teva between January 1, 2015 and April 24, 2015, or Closing Date. Teva was permitted to deduct from the $3.0 million hold-back certain costs incurred after January 1, 2015 that arose after the Closing Date. Teva is responsible for expenses related to custirsen incurred pursuant to the Collaboration Agreement through December 31, 2014. We are responsible for certain custirsen-related expenses from and after January 1, 2015. Pursuant to the Termination Agreement, we received a nominal amount from the remaining hold-back after deductions by Teva for certain costs incurred after the Closing Date. We do not expect to receive any additional amounts from Teva.
In accordance with the Termination Agreement, Teva transferred certain third-party agreements for the phase 3 clinical trial in second-line chemotherapy in patients with non-small cell lung cancer, or ENSPIRIT, and custirsen development activities to us on the Closing Date. If any additional historical third-party agreements are discovered after the Closing Date and are used to conduct the ENSPIRIT study, then Teva will use commercially reasonable effort to assign such agreements to us and will be responsible for any costs invoiced under such agreements in excess of an aggregate of $0.1 million. We will be responsible for the initial $0.1 million of costs under such agreements.
All licenses granted by us to Teva under the Collaboration Agreement were terminated as of the Closing Date. In addition, Teva assigned to us certain patent applications related to custirsen and abandoned certain other patent applications as requested by us. Furthermore, Teva granted to us and our affiliates an exclusive license (except as to Teva and its affiliates) to any know-how created under and during the term of the Collaboration Agreement to develop, manufacture and commercialize custirsen and certain other antisense inhibitors of clusterin, as set forth in more detail in the Termination Agreement. Teva additionally granted to us and our affiliates a non-exclusive license to any intellectual property owned by or licensed to Teva and its affiliates, whether as of the Closing Date or thereafter, to develop, manufacture and commercialize custirsen, subject to certain limitations. Teva also agreed not to challenge the patentability, validity or enforceability of certain of our patents, and agreed not to file any patent applications covering custirsen or any antisense inhibitor of clusterin for 18 months after the Closing Date. We are responsible for any such expenses incurred from and after January 1, 2015. We do not owe Teva any development milestone payments or royalty payments on sales of custirsen, if any.
As part of the termination, Teva assigned to us the investigational new drug application for custirsen and submitted amendments, on a country-by-country basis, transferring sponsorship of the ENSPIRIT study to us. In July 2015, we became the sole trial sponsor for the ENSPIRIT study in all countries.
We and Teva released each other from all claims related to the Collaboration Agreement. In addition, we agreed to indemnify Teva and its affiliates against any third-party claims attributable to the development and commercialization of custirsen prior to the
64
execution of the Collaboration Agreement and after the Closing Date, and any third-party claims attributable to the conduct of the phase 3 clinical trial in second-line chemotherapy in patients with metastatic castrate resistant prostate cancer, or AFFINITY. Teva agreed to indemnify us and our affiliates against any third-party claims attributable to the development of custirsen during the period between the execution of the Collaboration Agreement and the Closing Date, but excluding the AFFINITY study. The parties’ indemnity obligations cover, among other things, third-party claims brought by current or former patients in the relevant studies and patient product liability claims.
Revenue for the year ended December 31, 2016 was $5.1 million, which consists of recognition of deferred collaboration revenue representing our efforts in the development of custirsen. As of June 30, 2016, the full amount of the advanced reimbursement payment was recognized into collaboration revenue. The advanced reimbursement payment made by Teva, as part of the Termination Agreement, was deferred and recognized as collaboration revenue on a dollar for dollar basis as costs were incurred as part of the continuing research and development activities related to custirsen.
Ionis and UBC License Agreements
In January 2017, we discontinued further development of OGX-225. We provided a notice of discontinuance to Ionis, notifying them that we have discontinued development of OGX-225 resulting in termination of the license agreement related to this product candidate. We intend to also terminate the UBC license agreement related to OGX-225 provided that Ionis does not exercise its reversion rights within 90 days of the notice of discontinuance. If Ionis exercises its reversion rights related to OGX-225, we believe Ionis will assume the rights and obligations under the UBC license agreement.
In November 2016, we provided a notice of discontinuance to Ionis, or the Notice of Discontinuance, and a letter of termination to UBC, or the Letter of Termination, notifying the parties that we have discontinued development of custirsen, resulting in termination of all licensing agreements related to custirsen. We believe that all financial obligations, other than continuing mutual indemnification obligations and our requirement to pay for out-of-pocket patent expenses incurred up to the date of termination and for abandoning the custirsen patents and patent applications, under all agreements with Ionis and UBC, including the Ionis settlement agreement, are no longer owed and no further payments are due.
Under the license agreements with Ionis and UBC, we were required to pay royalties to each of Ionis and UBC based on a percentage of net sales. We did not make any royalty payments to either Ionis or UBC in 2016. In addition, pursuant to the terms of the agreements with Ionis, we were required to pay to Ionis up to 20% of all non-royalty revenue (defined to mean revenue not based on net sales of products) we receive from third parties.
In May and November 2015, we received communications from Ionis requesting payment of 30% of the $23.2 million paid by Teva under the Termination Agreement, as well as 30% of any amounts paid by Teva upon release of the $3.0 million holdback amount. In January 2016, Ionis filed a lawsuit and claimed that we were in breach of the license agreement for failing to pay Ionis a share of the advance reimbursement payment from Teva and other non-monetary consideration received from Teva in connection with the termination of the Collaboration Agreement. Ionis sought damages and a declaratory judgment that, based on our alleged breach, Ionis has the right to terminate the license agreement.
In August 2016, we and Ionis settled this lawsuit. Pursuant to the settlement, we paid to Ionis a $1.4 million upfront payment. In addition, under the settlement agreement, we were required to pay to Ionis additional success-based payments of up to an amount that does not exceed $5.0 million based on, (i) an additional 5% royalty on net sales of custirsen and (ii) 50% of any money we receive related to the sale, license or any other commercial transaction involving custirsen, subject to certain limitations. As a result of the Notice of Discontinuance, we believe that all financial obligations under the settlement agreement are no longer owed and no further payments are due.
5. FAIR VALUE MEASUREMENTS
Assets and liabilities recorded at fair value in the balance sheets are categorized based upon the level of judgment associated with the inputs used to measure their fair value. For certain of our financial instruments including amounts receivable and accounts payable the carrying values approximate fair value due to their short-term nature.
65
ASC 820 “Fair Value Measurements and Disclosures,” specifies a hierarchy of valuation techniques based on whether the inputs to those valuation techniques are observable or unobservable. In accordance with ASC 820, these inputs are summarized in the three broad level listed below:
|
|
• |
Level 1 – Quoted prices in active markets for identical securities. |
|
|
• |
Level 2 – Other significant inputs that are observable through corroboration with market data (including quoted prices in active markets for similar securities). |
|
|
• |
Level 3 – Significant unobservable inputs that reflect management’s best estimate of what market participants would use in pricing the asset or liability. |
As quoted prices in active markets are not readily available for certain financial instruments, we obtain estimates for the fair value of financial instruments through third-party pricing service providers.
In determining the appropriate levels, we performed a detailed analysis of the assets and liabilities that are subject to ASC 820.
We invest our excess cash in accordance with investment guidelines that limit the credit exposure to any one financial institution other than securities issued by the U.S. Government. These securities are not collateralized and mature within one year.
A description of the valuation techniques applied to our financial instruments measured at fair value on a recurring basis follows.
Financial Instruments
Cash
Significant amounts of cash are held on deposit with large well established U.S. and Canadian financial institutions.
U.S. Government and Agency Securities
U.S. Government Securities U.S. government securities are valued using quoted market prices. Valuation adjustments are not applied. Accordingly, U.S. government securities are categorized in Level 1 of the fair value hierarchy.
U.S. Agency Securities U.S. agency securities are comprised of two main categories consisting of callable and non-callable agency issued debt securities. Non-callable agency issued debt securities are generally valued using quoted market prices. Callable agency issued debt securities are valued by benchmarking model-derived prices to quoted market prices and trade data for identical or comparable securities. Actively traded non-callable agency issued debt securities are categorized in Level 1 of the fair value hierarchy. Callable agency issued debt securities are categorized in Level 2 of the fair value hierarchy.
Corporate and Other Debt
Corporate Bonds and Commercial Paper The fair value of corporate bonds and commercial paper is estimated using recently executed transactions, market price quotations (where observable), bond spreads or credit default swap spreads adjusted for any basis difference between cash and derivative instruments. The spread data used are for the same maturity as the bond. If the spread data does not reference the issuer, then data that reference a comparable issuer are used. When observable price quotations are not available, fair value is determined based on cash flow models with yield curves, bond or single name credit default swap spreads and recovery rates based on collateral values as significant inputs. Corporate bonds and commercial paper are generally categorized in Level 2 of the fair value hierarchy; in instances where prices, spreads or any of the other aforementioned key inputs are unobservable, they are categorized in Level 3 of the hierarchy.
Warrants
As of December 31, 2016, we recorded a $0.2 million warrant liability. We reassess the fair value of the common stock warrants classified as liabilities at each reporting date utilizing a Black-Scholes pricing model. Inputs used in the pricing model include estimates of stock price volatility, expected warrant life and risk-free interest rate. The computation of expected volatility is based on the historical volatility of shares of our common stock for a period that coincides with the expected life of the warrants that are classified as liabilities. Warrants that are classified as liabilities are categorized in Level 3 of the fair value hierarchy. A small change in the estimates used may have a relatively large change in the estimated valuation. Warrants that are classified as equity are not considered liabilities and therefore are not reassessed for their fair values at each reporting date.
66
The following table presents the changes in fair value of our total Level 3 financial liabilities for the year ended December 31, 2016. During the twelve months ended December 31, 2016, no common stock warrants were issued that were classified as liabilities (in thousands):
|
|
|
Liability at |
|
|
|
|
|
|
Unrealized |
|
|
Liability at |
|
|||
|
|
|
December 31, |
|
|
Issuance of |
|
|
Gain on |
|
|
December 31, |
|
||||
|
|
|
2015 |
|
|
Warrants |
|
|
warrants |
|
|
2016 |
|
||||
|
Warrant liability |
|
$ |
1,105 |
|
|
$ |
— |
|
|
$ |
(873 |
) |
|
$ |
232 |
|
The following table presents information about our assets and liabilities that are measured at fair value on a recurring basis, and indicates the fair value hierarchy of the valuation techniques we utilized to determine such fair value (in thousands):
|
December 31, 2016 |
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
||||
|
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash |
|
$ |
1,800 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
1,800 |
|
|
Money market securities (cash equivalents) |
|
|
11,931 |
|
|
|
— |
|
|
|
— |
|
|
|
11,931 |
|
|
Corporate bonds and commercial paper (cash equivalents) |
|
|
1,502 |
|
|
|
— |
|
|
|
— |
|
|
|
1,502 |
|
|
Government securities |
|
|
2,000 |
|
|
|
— |
|
|
|
— |
|
|
|
2,000 |
|
|
Restricted cash (Note 12) |
|
|
272 |
|
|
|
— |
|
|
|
— |
|
|
|
272 |
|
|
Corporate bonds and commercial paper (short term investments) |
|
|
— |
|
|
|
8,230 |
|
|
|
— |
|
|
|
8,230 |
|
|
Total assets |
|
$ |
17,505 |
|
|
$ |
8,230 |
|
|
$ |
— |
|
|
$ |
25,735 |
|
|
Liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Warrants |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
232 |
|
|
$ |
232 |
|
|
December 31, 2015 |
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
||||
|
Assets |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash |
|
$ |
14,034 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
14,034 |
|
|
Money market securities (cash equivalents) |
|
|
20,276 |
|
|
|
— |
|
|
|
— |
|
|
|
20,276 |
|
|
Restricted cash (Note 12) |
|
|
272 |
|
|
|
— |
|
|
|
— |
|
|
|
272 |
|
|
Corporate bonds and commercial paper |
|
|
— |
|
|
|
20,876 |
|
|
|
— |
|
|
|
20,876 |
|
|
Total assets |
|
$ |
34,582 |
|
|
$ |
20,876 |
|
|
$ |
— |
|
|
$ |
55,458 |
|
|
Liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Warrants |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
1,105 |
|
|
$ |
1,105 |
|
Cash and cash equivalents and short term investments (in thousands):
|
|
|
|
|
|
|
Gross |
|
|
Gross |
|
|
|
|
|
||
|
|
|
Amortized |
|
|
Unrealized |
|
|
Unrealized |
|
|
Estimated |
|
||||
|
December 31, 2016 |
|
Cost |
|
|
Gains |
|
|
Losses |
|
|
Fair Value |
|
||||
|
Cash |
|
$ |
1,800 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
1,800 |
|
|
Money market securities |
|
|
11,931 |
|
|
|
— |
|
|
|
— |
|
|
|
11,931 |
|
|
Corporate bonds and commercial paper |
|
|
1,502 |
|
|
|
— |
|
|
|
— |
|
|
|
1,502 |
|
|
Total cash and cash equivalents |
|
$ |
15,233 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
15,233 |
|
|
Money market securities (restricted cash) |
|
|
272 |
|
|
|
— |
|
|
|
— |
|
|
|
272 |
|
|
Total restricted cash |
|
$ |
272 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
272 |
|
|
Corporate bonds and commercial paper |
|
|
8,231 |
|
|
|
— |
|
|
|
(1 |
) |
|
|
8,230 |
|
|
Government securities |
|
|
2,000 |
|
|
|
— |
|
|
|
— |
|
|
|
2,000 |
|
|
Total short-term investments |
|
$ |
10,231 |
|
|
$ |
— |
|
|
$ |
(1 |
) |
|
$ |
10,230 |
|
67
|
|
|
|
|
|
|
Gross |
|
|
Gross |
|
|
|
|
|
||
|
|
|
Amortized |
|
|
Unrealized |
|
|
Unrealized |
|
|
Estimated |
|
||||
|
December 31, 2015 |
|
Cost |
|
|
Gains |
|
|
Losses |
|
|
Fair Value |
|
||||
|
Cash |
|
$ |
14,034 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
14,034 |
|
|
Money market securities |
|
|
20,276 |
|
|
|
— |
|
|
|
— |
|
|
|
20,276 |
|
|
Total cash and cash equivalents |
|
$ |
34,310 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
34,310 |
|
|
Money market securities (restricted cash) |
|
|
272 |
|
|
|
— |
|
|
|
— |
|
|
|
272 |
|
|
Total restricted cash |
|
$ |
272 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
272 |
|
|
Corporate bonds and commercial paper |
|
|
20,885 |
|
|
|
— |
|
|
|
(9 |
) |
|
|
20,876 |
|
|
Total short-term investments |
|
$ |
20,885 |
|
|
$ |
— |
|
|
$ |
(9 |
) |
|
$ |
20,876 |
|
Our gross realized gains and losses on sales of available-for-sale securities were not material for the years ended December 31, 2016 and 2015.
All securities included in cash and cash equivalents have maturities of 90 days or less at the time of purchase. All securities included in short-term investments have maturities of within one year of the balance sheet date. The cost of securities sold is based on the specific identification method.
We only invest in A (or equivalent) rated securities with maturities of one year or less. We do not believe that there are any other than temporary impairments related to our investment in marketable securities at December 31, 2016, given the quality of the investment portfolio, its short-term nature, and subsequent proceeds collected on sale of securities that reached maturity.
6. PROPERTY AND EQUIPMENT
Property and equipment consisted of the following (in thousands):
|
|
|
|
|
|
|
Accumulated |
|
|
Net Book |
|
||
|
|
|
Cost |
|
|
Depreciation |
|
|
Value |
|
|||
|
December 31, 2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Computer equipment |
|
$ |
657 |
|
|
$ |
567 |
|
|
$ |
90 |
|
|
Furniture and fixtures |
|
|
172 |
|
|
|
167 |
|
|
|
5 |
|
|
Machinery and equipment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Leasehold improvements |
|
|
262 |
|
|
|
167 |
|
|
|
95 |
|
|
Computer software |
|
|
502 |
|
|
|
439 |
|
|
|
63 |
|
|
Equipment under capital lease |
|
|
114 |
|
|
|
109 |
|
|
|
5 |
|
|
Total property and equipment |
|
$ |
1,707 |
|
|
$ |
1,449 |
|
|
$ |
258 |
|
|
December 31, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Computer equipment |
|
$ |
628 |
|
|
$ |
490 |
|
|
$ |
138 |
|
|
Furniture and fixtures |
|
|
172 |
|
|
|
163 |
|
|
|
9 |
|
|
Machinery and equipment |
|
|
218 |
|
|
|
1 |
|
|
|
217 |
|
|
Leasehold improvements |
|
|
257 |
|
|
|
99 |
|
|
|
158 |
|
|
Computer software |
|
|
494 |
|
|
|
425 |
|
|
|
69 |
|
|
Equipment under capital lease |
|
|
114 |
|
|
|
103 |
|
|
|
11 |
|
|
Total property and equipment |
|
$ |
1,883 |
|
|
$ |
1,281 |
|
|
$ |
602 |
|
Impairment of Long-Lived Assets
We review long-lived assets for impairment whenever events or changes in circumstances indicate that the asset’s carrying amount may not be recoverable. We conduct our long-lived asset impairment analyses in accordance with ASC 360-10-15, “Impairment or Disposal of Long-Lived Assets.” ASC 360-10-15 requires us to group assets and liabilities at the lowest level for which identifiable cash flows are largely independent of the cash flows of other assets and liabilities and evaluate the asset group against the sum of the undiscounted future cash flows. If the undiscounted cash flows do not indicate the carrying amount of the asset is recoverable, an impairment charge is measured as the amount by which the carrying amount of the asset group exceeds its fair value based on discounted cash flow analysis or appraisals.
68
In the fourth quarter of 2016, we concluded that we had a triggering event requiring assessment of impairment for certain of our long-lived assets in conjunction with our restructuring actions announced in October 2016. As a result, we reviewed our long-lived assets for impairment and recorded a $0.2 million impairment charge, representing the entire amount of the then carrying value of the machinery and equipment, on our statement of loss. The full amount of the impairment charge related to drug product manufacturing equipment.
7. EXCESS LEASE LIABILITY
On August 21, 2008, Sonus Pharmaceuticals, Inc., or Sonus, completed a transaction (“the Arrangement”) with OncoGenex Technologies Inc., or OncoGenex Technologies, whereby Sonus acquired all of the outstanding preferred shares, common shares and convertible debentures of OncoGenex Technologies. Sonus then changed its name to OncoGenex Pharmaceuticals, Inc. Prior to the Arrangement, Sonus entered into a non-cancellable lease arrangement for office space located in Bothell, Washington, which is considered to be in excess of our current requirements. The liability was computed as the present value of the difference between the remaining lease payments due less the estimate of net sublease income and expenses and had been accounted for in accordance with ASC 805-20, “Business Combinations -Identifiable Assets and Liabilities, and Any Non-controlling Interest.” Effective 2014, we entered into a Lease Termination Agreement with the landlord for the office space in Bothell such that the lease terminated effective March 1, 2015. Under the Lease Termination Agreement, we paid BMR-217TH Place LLC (“BMR”) a $2.0 million termination fee on the Termination Date. We agreed to pay BMR an additional termination fee of $1.3 million within 30 days after we (i) meet the primary endpoint for our phase 3 clinical trial for the treatment of second line metastatic CRPC with custirsen and (ii) close a transaction or transactions pursuant to which we receive funding in an aggregate amount of at least $20.0 million. As a result of the Lease Termination Agreement, we have recorded the lease termination fees and have made an adjustment to remove the excess lease liability. We re-assessed that the likelihood of meeting both contingent events is no longer possible due to not achieving the primary endpoint on our AFFINITY trial. As a result, we have reversed the $1.3 million in lease termination liability on our balance sheet during the third quarter of 2016 and recognized a recovery on our statement of loss.
8. OTHER ASSETS
Other assets include prepaid amounts related to clinical trials that will not be utilized in the next 12 months and deposits paid for office space in accordance with the terms of the operating lease agreements.
9. INCOME TAX
[a] The reconciliation of income tax attributable to operations computed at the statutory tax rate to income tax expense is as follows. OncoGenex Technologies, a Canadian corporation, which is subject to combined Canadian federal and provincial statutory tax rates for December 31, 2016, 2015, and 2014 of 26.0%, 26.0%, and 26.0%, respectively. Following the reverse takeover by OncoGenex Technologies of Sonus Pharmaceuticals, Inc. (which subsequently changed its name to OncoGenex Pharmaceuticals, Inc.) in 2008, OncoGenex Technologies became a wholly owned subsidiary of OncoGenex Pharmaceuticals, which is a Delaware incorporated company subject to US Federal Statutory rates of 34% for all three years presented.
For the purposes of estimating the tax rate in effect at the time that deferred tax assets and liabilities are expected to reverse, we used the furthest out available future tax rate in the applicable jurisdictions. For the years ended December 31, 2016, 2015 and 2014 the future Canadian enacted rates we used were 26%, 26%, and 26%, respectively, while for the US the future enacted rate we used was 34% for all three periods presented.
[b] At December 31, 2016, we have investment tax credits of $2.6 million (2015—$2.3 million) available to reduce future Canadian income taxes otherwise payable. We also have non-capital loss carryforwards of $115.9 million (2015—$100.4 million) available to offset future taxable income in Canada and federal net operating loss carryforwards of $158.4 million (2015—$151.9 million) to offset future taxable income in the United States.
69
Under Section 382 of the Internal Revenue Code of 1986, substantial changes in our ownership may limit the amount of net operating loss carryforwards and development tax credit carryforwards that could be utilized annually in the future to offset taxable income. Any such annual limitation may significantly reduce the utilization of the net operating losses and tax credits before they expire. A preliminary 382 limitation review has been undertaken but a formal study has never been completed. The results of any future study could indicate that the U.S. losses may be materially limited; however, the amount of such limitation cannot be reasonably quantified at this time, but may be significant. In each period since our inception, we have recorded a valuation allowance for the full amount of our deferred tax asset, as the realization of the deferred tax asset is uncertain.
|
(In thousands) |
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Income taxes at statutory rates (at a rate of 34% for all periods presented) |
|
$ |
(6,844 |
) |
|
$ |
(5,712 |
) |
|
$ |
(8,922 |
) |
|
Expenses not deducted for tax purposes |
|
|
(67 |
) |
|
|
(14 |
) |
|
|
(452 |
) |
|
Effect of tax rate changes on deferred tax assets and liabilities |
|
|
(3 |
) |
|
|
(13 |
) |
|
|
(9 |
) |
|
Rate differential on foreign earnings |
|
|
972 |
|
|
|
689 |
|
|
|
1,445 |
|
|
Reduction (increase) in benefit of operating losses |
|
|
196 |
|
|
|
(32 |
) |
|
|
441 |
|
|
Reduction in the benefit of other tax attributes |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Investment tax credits |
|
|
(252 |
) |
|
|
(297 |
) |
|
|
(357 |
) |
|
Change in valuation allowance |
|
|
6,203 |
|
|
|
5,114 |
|
|
|
7,854 |
|
|
Book to tax return adjustments |
|
|
(205 |
) |
|
|
265 |
|
|
|
— |
|
|
Other |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
Income tax expense |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
As a result, we have not recognized any federal or state income tax benefit in our statement of operations. The initial public offering of common stock by us in 1995 caused an ownership change pursuant to applicable regulations in effect under the Internal Revenue Code of 1986. Therefore, our use of losses incurred through the date of ownership change will be limited during the carryforward period and may result in the expiration of net operating loss carryforwards in the United States before utilization.
The investment tax credits and non-capital losses and net operating losses for income tax purposes expire as follows (in thousands):
|
|
|
Investment |
|
|
Net Operating |
|
|
Non-capital |
|
|||
|
|
|
Tax Credits |
|
|
Losses |
|
|
Losses |
|
|||
|
2016 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
2017 |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
2018 |
|
|
150 |
|
|
|
10,795 |
|
|
|
— |
|
|
2019 |
|
|
102 |
|
|
|
32 |
|
|
|
— |
|
|
2020 |
|
|
76 |
|
|
|
2,745 |
|
|
|
— |
|
|
2021 |
|
|
69 |
|
|
|
400 |
|
|
|
— |
|
|
2022 |
|
|
105 |
|
|
|
11,766 |
|
|
|
— |
|
|
2023 |
|
|
96 |
|
|
|
10,785 |
|
|
|
— |
|
|
2024 |
|
|
111 |
|
|
|
16,814 |
|
|
|
— |
|
|
2025 |
|
|
144 |
|
|
|
2,062 |
|
|
|
— |
|
|
2026 |
|
|
400 |
|
|
|
27,157 |
|
|
|
7,335 |
|
|
2027 |
|
|
173 |
|
|
|
22,225 |
|
|
|
4,949 |
|
|
2028 |
|
|
390 |
|
|
|
12,648 |
|
|
|
8,020 |
|
|
2029 |
|
|
317 |
|
|
|
4,358 |
|
|
|
(9 |
) |
|
2030 |
|
|
346 |
|
|
|
5,034 |
|
|
|
6,288 |
|
|
2031 |
|
|
608 |
|
|
|
6,200 |
|
|
|
12,121 |
|
|
2032 |
|
|
505 |
|
|
|
8,418 |
|
|
|
17,278 |
|
|
2033 |
|
|
411 |
|
|
|
2,366 |
|
|
|
23,240 |
|
|
2034 |
|
|
492 |
|
|
|
2,609 |
|
|
|
17,077 |
|
|
2035 |
|
|
328 |
|
|
|
5,342 |
|
|
|
3,120 |
|
|
2036 |
|
|
286 |
|
|
|
6,635 |
|
|
|
16,531 |
|
|
|
|
$ |
5,109 |
|
|
$ |
158,391 |
|
|
$ |
115,950 |
|
70
In addition, we have unclaimed tax deductions of approximately $14.6 million related to scientific research and experimental development expenditures available to carry forward indefinitely to reduce Canadian taxable income of future years. We also have research and development tax credits of $2.4 million available to reduce future taxes payable in the United States. The research and development tax credits expire between 2018 and 2036.
[c] Significant components of our deferred tax assets as of December 31 are shown below (in thousands):
The potential income tax benefits relating to these deferred tax assets have not been recognized in the accounts as their realization did not meet the requirements of “more likely than not” under the liability method of tax allocation. Accordingly, a valuation allowance has been recorded and no deferred tax assets have been recognized as at December 31, 2016 and 2015.
|
|
|
2016 |
|
|
2015 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
Tax basis in excess of book value of assets |
|
$ |
6,483 |
|
|
$ |
6,308 |
|
|
Non-capital loss carryforwards |
|
|
84,358 |
|
|
|
77,746 |
|
|
Research and development deductions and credits |
|
|
7,969 |
|
|
|
7,484 |
|
|
Stock options |
|
|
3,743 |
|
|
|
3,448 |
|
|
Restructuring liability |
|
|
624 |
|
|
|
474 |
|
|
Other |
|
|
112 |
|
|
|
1,627 |
|
|
Total deferred tax assets |
|
|
103,289 |
|
|
|
97,087 |
|
|
Valuation allowance |
|
$ |
(103,289 |
) |
|
$ |
(97,087 |
) |
|
|
|
|
|
|
|
|
|
|
|
Net deferred tax assets |
|
|
- |
|
|
|
- |
|
[d] Under ASC 740, the benefit of an uncertain tax position that is more likely than not of being sustained upon audit by the relevant taxing authority must be recognized at the largest amount that is more likely than not to be sustained. No portion of the benefit of an uncertain tax position may be recognized if the position has less than a 50% likelihood of being sustained.
A reconciliation of the unrecognized tax benefits of uncertain tax positions for the year ended December 31, 2016 is as follows (in thousands):
|
|
|
Year ended |
|
|||||||||
|
|
|
December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Balance at January 1 |
|
$ |
2,055 |
|
|
$ |
2,039 |
|
|
$ |
2,007 |
|
|
Additions based on tax positions related to the current year |
|
|
16 |
|
|
|
16 |
|
|
|
32 |
|
|
Additions based on tax positions related to prior years |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Balance at December 31 |
|
$ |
2,071 |
|
|
$ |
2,055 |
|
|
$ |
2,039 |
|
As of December 31, 2016, unrecognized benefits of approximately $2.0 million, if recognized, would affect our effective tax rate, and would reduce our deferred tax assets.
Our accounting policy is to treat interest and penalties relating to unrecognized tax benefits as a component of income taxes. As of December 31, 2016 and December 31, 2015 we had no accrued interest and penalties related to income taxes.
We are subject to taxes in Canada and the U.S. until the applicable statute of limitations expires. Tax audits by their very nature are often complex and can require several years to complete.
|
Tax |
|
Years open to |
|
Jurisdiction |
|
examination |
|
Canada |
|
2008 to 2016 |
|
US |
|
2013 to 2016 |
10. COMMON STOCK
[a] Authorized
75,000,000 authorized common voting share, par value of $0.001, and 5,000,000 preferred shares, par value of $0.001.
71
[b] Issued and outstanding shares
At-The-Market Issuance Sales Agreement
In June 2013, we entered into an At-the-Market Issuance Sales Agreement, or Sales Agreement, with MLV & Co. LLC, or MLV, under which we may offer and sell shares of our common stock having aggregate sales proceeds of up to $25 million from time to time through MLV as our sales agent. Sales of our common stock through MLV, if any, will be made by any method permitted that is deemed an “at the market” offering as defined in Rule 415 under the Securities Act of 1933, as amended, including by means of ordinary brokers’ transactions on The NASDAQ Capital Market or otherwise at market prices prevailing at the time of sale, in block transactions, or as otherwise agreed upon by us and MLV. MLV will use commercially reasonable efforts to sell our common stock from time to time, based upon instructions from us (including any price, time or size limits or other customary parameters or conditions we may impose). We will pay MLV a commission of up to 3.0% of the gross sales proceeds of any shares of common stock sold through MLV under the Sales Agreement. We are not obligated to make any sales of common stock under the Sales Agreement. The offering of our shares of common stock pursuant to the Sales Agreement will terminate upon the earlier of (i) the sale of all common stock subject to the Sales Agreement or (ii) termination of the Sales Agreement in accordance with its terms.
On April 27, 2015, we and MLV terminated the Sales Agreement. We were not subject to any termination penalties related to termination of the Sales Agreement. Under the Sales Agreement, we offered and sold 809,214 shares of our common stock with MLV & Co. LLC. These sales resulted in gross proceeds to us of approximately $3.0 million and offering expenses of $0.1 million.
July 2014 Registered Offering
On July 2, 2014, we completed an underwritten registered offering pursuant to which we sold 5,559,866 Series A units at a price per unit of $3.48 and 1,340,538 Series B units at a price per unit of $3.47.
Each Series A unit consisted of one share of common stock and a Series A warrant to purchase up to one-half of one share of common stock at an initial exercise price of $4.00 per share. Each Series A warrant is exercisable at any time on or after the date of issuance until the fifth anniversary of the issuance of the Series A warrants.
Each Series B unit consisted of a Pre-Funded Series B warrant to purchase up to one share of common stock at an initial exercise price of $0.01 per share and a Series B warrant to purchase up to one-half of one share of common stock at an initial exercise price of $4.00 per share. Each Pre-Funded Series B warrant and Series B warrant is exercisable at any time on or after the date of issuance until the fifth anniversary of the issuance of the Pre-Funded Series B warrants and Series B warrants, respectively.
We received net proceeds of approximately $22.4 million, after deducting underwriting discounts and commissions and offering expenses. Gross proceeds of $24.0 million and underwriting discounts and commissions and offering expenses of $1.6 million were allocated as follows:
|
|
|
|
|
|
|
Series B |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Pre-funded |
|
|
Series A |
|
|
Series B |
|
|||
|
|
|
|
|
|
|
Common |
|
|
Common |
|
|
Common |
|
|||
|
|
|
Common |
|
|
Stock |
|
|
Stock |
|
|
Stock |
|
||||
|
|
|
Stock |
|
|
Warrants |
|
|
Warrants |
|
|
Warrants |
|
||||
|
Units Issued |
|
|
5,559,866 |
|
|
|
1,340,538 |
|
|
|
2,779,933 |
|
|
|
670,269 |
|
|
Gross Proceeds (in thousands) |
|
$ |
14,084 |
|
|
$ |
3,387 |
|
|
$ |
5,261 |
|
|
$ |
1,268 |
|
|
Underwriting discount and offering expense (in thousands) |
|
$ |
885 |
|
|
$ |
213 |
|
|
$ |
428 |
|
|
$ |
103 |
|
The Series A and Series B common stock warrants are classified as liabilities. The underwriting discount and offering expenses allocated to the Series A and Series B common stock warrants have been expensed in the Consolidated Statement of Loss.
The common stock and Series B prefunded common stock warrants are classified as equity. The underwriting discount and offering expenses allocated to the common stock and Series B prefunded common stock warrants have been charged against the allocated gross proceeds.
Purchase Agreement and Financing with Lincoln Park Capital
On April 30, 2015, we and Lincoln Park Capital Fund, LLC, or LPC, entered into a share and unit purchase agreement, or Purchase Agreement, pursuant to which we have the right to sell to LPC up to $18.0 million in shares of our common stock, par value $0.001 per share, subject to certain limitations and conditions set forth in the Purchase Agreement.
72
Pursuant to the Purchase Agreement, LPC initially purchased 956,938 Series A-1 Units at a purchase price of $2.09 per unit, with each Series A-1 Unit consisting of (a) one share of Common Stock and (b) one warrant to purchase one-quarter of a share of Common Stock at an exercise price of $2.40 per share, or Series A-1 Warrant. Each Series A-1 Warrant is exercisable six months following the issuance date until the date that is five years and six months after the issuance date and is subject to customary adjustments. The Series A-1 Warrants were issued only as part of the Series A-1 Units in the initial purchase of $2.0 million and no warrants were issued in connection with any other purchases of common stock under the Purchase Agreement.
After the initial purchase, as often as every business day over the 24-month term of the Purchase Agreement, and up to an aggregate amount of an additional $16.0 million (subject to certain limitations) of shares of common stock, we had the right, from time to time, in our sole discretion and subject to certain conditions to direct LPC to purchase up to 125,000 shares of common stock with such amounts increasing as the closing sale price of our common stock as reported on The NASDAQ Capital Market increased. The purchase price of shares of common stock pursuant to the Purchase Agreement was based on prevailing market prices of common stock at the time of sales without any fixed discount, and we controlled the timing and amount of common stock sold to LPC. In addition, we had the right to direct LPC to purchase additional amounts as accelerated purchases if on the date of a regular purchase the closing sale price of the common stock is not below $1.50 per share. As consideration for entering into the Purchase Agreement, we issued to LPC 126,582 shares of common stock; no cash proceeds were received from the issuance of these shares.
From April 30, 2015 through August 13, 2015, we offered and sold 6,814,980 shares of our common stock pursuant to our Purchase Agreement with LPC. These sales resulted in gross proceeds to us of approximately $18.0 million and offering expenses of $0.4 million. As of August 13, 2015, no further amounts remained available for sale under this offering program
Stock Option Exercises
During the year ended December 31, 2016, we did not issue any shares of common stock to satisfy stock option exercises and issued 217,296 shares of common stock to satisfy and restricted stock unit settlements, respectively, compared with the issuance of 5,359 and 269,401 shares of common to satisfy stock option exercises and restricted stock unit settlements, respectively, for the years ended December 31, 2015. For the year ended December 31, 2014, we issued 10,000 and 203,148 shares of common stock to satisfy stock option exercises and restricted stock unit settlements, respectively.
[c] Stock options
As at December 31, 2016 we had reserved, pursuant to our 2010 Performance Incentive Plan, 3,634,058 common shares for issuance upon exercise of stock options and settlement of restricted stock units by employees, directors, officers and consultants of ours, of which 1,378,805 are reserved for options currently outstanding, 253,221 are reserved for restricted stock units currently outstanding and 2,002,032 are available for future equity award grants under our 2010 Performance Incentive Plan. As of December 31, 2015 3,876,151 shares were available for equity award grants under our 2010 Performance Incentive Plan.
2010 Performance Incentive Plan
At our 2013 Annual Meeting of Stockholders held on May 24, 2013, our stockholders approved an amendment to our 2010 Performance Incentive Plan, or the 2010 Plan. As a result of this amendment, the 2010 Plan was further amended to provide for an increase in the total shares of common stock available for issuance under the 2010 Plan from 1,050,000 to 2,050,000. At our 2014 Annual Meeting of Stockholders held on May 29, 2014, our stockholders approved an amendment to our 2010 Performance Incentive Plan. As a result of this amendment, the 2010 Plan was amended to provide for an increase in the total shares of common stock available for issuance under the 2010 Plan from 2,050,000 to 2,800,000. At our 2015 Annual Meeting of Stockholders held on May 21, 2015, our stockholders approved an amendment to our 2010 Performance Incentive Plan. As a result of this amendment, the 2010 Plan was amended to provide for an increase in the total shares of common stock available for issuance under the 2010 Plan from 2,800,000 to 4,300,000.
Under the plan, we may grant options to purchase common shares or restricted stock units to our employees, directors, officers and consultants. The exercise price of the options is determined by our board of directors but will be at least equal to the fair value of the common shares at the grant date. The options vest in accordance with terms as determined by our board of directors, typically over three to four years for options issued to employees and consultants, and over one to three years for members of our board of directors. The expiry date for each option is set by our board of directors with a maximum expiry date of ten years from the date of grant. In addition, the 2010 Plan allows for accelerated vesting of outstanding equity awards in the event of a change in control. The terms for accelerated vesting, in the event of a change in control, is determined at our discretion and defined under the employment agreements for our officers and certain of our employees.
73
Options remain outstanding under a number of share option plans that had been approved by shareholders prior to the approval of the 2010 Performance Incentive Plan: (a) the 2007 Performance Incentive Plan (2007 Plan).
ASC 718 Compensation – Stock Compensation
We recognize expense related to the fair value of our stock-based compensation awards using the provisions of ASC 718. We use the Black-Scholes option pricing model as the most appropriate fair value method for our stock options and recognize compensation expense for stock options on a straight-line basis over the requisite service period. In valuing our stock options using the Black-Scholes option pricing model, we make assumptions about risk-free interest rates, dividend yields, volatility and weighted average expected lives, including estimated forfeiture rates of the options.
The expected life was calculated based on the simplified method as permitted by the SEC’s Staff Accounting Bulletin 110, Share-Based Payment. We consider the use of the simplified method appropriate because we believe our historical stock option exercise activity may not be indicative of future stock option exercise activity based upon the structural changes to our business that may occur as a result of merger with Achieve Life Science, Inc. and the potential impact on future stock option exercise activity. The expected volatility of options granted was calculated based on the historical volatility of the shares of our common stock. The risk-free interest rate is based on a U.S. Treasury instrument whose term is consistent with the expected life of the stock options. In addition to the assumptions above, as required under ASC 718, management made an estimate of expected forfeitures and is recognizing compensation costs only for those equity awards expected to vest. Forfeiture rates are estimated using historical actual forfeiture rates. These rates are adjusted on a quarterly basis and any change in compensation expense is recognized in the period of the change. We have never paid or declared cash dividends on our common stock and do not expect to pay cash dividends in the foreseeable future.
The estimated fair value of stock options granted in the respective periods was determined using the Black-Scholes option pricing model using the following weighted average assumptions:
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Risk-free interest rates |
|
|
1.51 |
% |
|
|
1.76 |
% |
|
|
1.83 |
% |
|
Expected dividend yield |
|
|
0 |
% |
|
|
0 |
% |
|
|
0 |
% |
|
Expected life |
|
5.3 years |
|
|
5.8 years |
|
|
5.9 years |
|
|||
|
Expected volatility |
|
|
72 |
% |
|
|
63 |
% |
|
|
82 |
% |
The weighted average fair value of stock options granted during the year ended December 31, 2016, 2015 and 2014 was $0.53, $1.10 and $6.52 per share, respectively.
The results for the periods set forth below included stock-based compensation expense in the following expense categories of the consolidated statements of loss (in thousands):
|
|
|
Years ended |
|
|||||||||
|
|
|
December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Research and development |
|
$ |
803 |
|
|
$ |
1,111 |
|
|
$ |
1,893 |
|
|
General and administrative |
|
|
846 |
|
|
|
1,217 |
|
|
|
1,967 |
|
|
Total stock-based compensation |
|
$ |
1,649 |
|
|
$ |
2,328 |
|
|
$ |
3,860 |
|
Options vest in accordance with terms as determined by our board of directors, typically over three or four years for employee and consultant grants and over one or three years for board of director option grants. The expiry date for each option is set by our board of directors with, which is typically seven to ten years. The exercise price of the options is determined by our board of directors but is at least equal to the fair value of the share at the grant date.
74
Stock option transactions and the number of stock options outstanding are summarized below:
|
|
|
Number of |
|
|
Weighted |
|
||
|
|
|
Optioned |
|
|
Average |
|
||
|
|
|
Common |
|
|
Exercise |
|
||
|
|
|
Shares |
|
|
Price |
|
||
|
Balance, January 1, 2014 |
|
|
1,007,491 |
|
|
$ |
11.39 |
|
|
Granted |
|
|
382,097 |
|
|
|
8.97 |
|
|
Expired |
|
|
(12,169 |
) |
|
|
18.93 |
|
|
Exercised |
|
|
(10,000 |
) |
|
|
3.00 |
|
|
Forfeited |
|
|
(84,000 |
) |
|
|
12.14 |
|
|
Balance, December 31, 2014 |
|
|
1,283,419 |
|
|
$ |
10.55 |
|
|
Granted |
|
|
502,047 |
|
|
|
1.91 |
|
|
Expired |
|
|
(247,766 |
) |
|
|
2.96 |
|
|
Exercised |
|
|
(5,359 |
) |
|
|
2.69 |
|
|
Forfeited |
|
|
(53,120 |
) |
|
|
14.37 |
|
|
Balance, December 31, 2015 |
|
|
1,479,221 |
|
|
$ |
8.78 |
|
|
Granted |
|
|
1,635,250 |
|
|
|
0.86 |
|
|
Expired |
|
|
(994,059 |
) |
|
|
1.00 |
|
|
Exercised |
|
|
— |
|
|
|
— |
|
|
Forfeited |
|
|
(741,607 |
) |
|
|
1.96 |
|
|
Balance, December 31, 2016 |
|
|
1,378,805 |
|
|
$ |
8.62 |
|
The following table summarizes information about stock options outstanding at December 31, 2016 regarding the number of ordinary shares issuable upon: (1) outstanding options and (2) vested options.
(1) Number of common shares issuable upon exercise of outstanding options:
|
|
|
|
|
|
|
|
|
|
|
Weighted- |
|
|
|
|
|
|
|
|
|
|
|
|
|
Average |
|
|
|
|
|
|
|
|
|
|
|
|
|
Remaining |
|
|
|
|
|
|
|
|
|
Weighted- |
|
|
Contractual |
|
||
|
|
|
|
|
|
|
Average |
|
|
Life |
|
||
|
Exercise Prices |
|
Number of Options |
|
|
Exercise Price |
|
|
(in years) |
|
|||
|
$1.00 - $1.54 |
|
|
122,010 |
|
|
$ |
1.00 |
|
|
|
9.36 |
|
|
$1.55 - $1.88 |
|
|
337,298 |
|
|
|
1.86 |
|
|
|
7.96 |
|
|
$1.89 - $3.40 |
|
|
118,874 |
|
|
|
2.62 |
|
|
|
6.98 |
|
|
$3.41 - $11.70 |
|
|
78,447 |
|
|
|
7.70 |
|
|
|
5.72 |
|
|
$11.71 - $11.86 |
|
|
185,105 |
|
|
|
11.79 |
|
|
|
6.66 |
|
|
$11.87 - $11.99 |
|
|
144,699 |
|
|
|
11.95 |
|
|
|
5.64 |
|
|
$12.00- $13.08 |
|
|
132,650 |
|
|
|
12.88 |
|
|
|
4.97 |
|
|
$13.09 - $16.40 |
|
|
135,576 |
|
|
|
15.64 |
|
|
|
3.54 |
|
|
$16.41 - $21.67 |
|
|
63,446 |
|
|
|
17.74 |
|
|
|
3.56 |
|
|
$21.68 - $22.28 |
|
|
60,700 |
|
|
|
22.28 |
|
|
|
2.67 |
|
|
|
|
|
1,378,805 |
|
|
$ |
8.62 |
|
|
|
6.30 |
|
75
(2) Number common shares issuable upon exercise of vested options:
|
|
|
|
|
|
|
|
|
|
|
Weighted- |
|
|
|
|
|
|
|
|
|
|
|
|
|
Average |
|
|
|
|
|
|
|
|
|
|
|
|
|
Remaining |
|
|
|
|
|
|
|
|
|
Weighted- |
|
|
Contractual |
|
||
|
|
|
|
|
|
|
Average |
|
|
Life |
|
||
|
Exercise Prices |
|
Number of Options |
|
|
Exercise Price |
|
|
(in years) |
|
|||
|
$1.00 - $1.54 |
|
|
510 |
|
|
$ |
1.21 |
|
|
|
0.42 |
|
|
$1.55 - $1.88 |
|
|
184,008 |
|
|
|
1.86 |
|
|
|
7.74 |
|
|
$1.89 - $3.40 |
|
|
90,275 |
|
|
|
2.51 |
|
|
|
6.76 |
|
|
$3.41 - $11.70 |
|
|
75,217 |
|
|
|
7.69 |
|
|
|
5.78 |
|
|
$11.71 - $11.86 |
|
|
140,269 |
|
|
|
11.79 |
|
|
|
6.58 |
|
|
$11.87 - $11.99 |
|
|
141,951 |
|
|
|
11.95 |
|
|
|
5.64 |
|
|
$12.00- $13.08 |
|
|
132,650 |
|
|
|
12.88 |
|
|
|
4.97 |
|
|
$13.09 - $16.40 |
|
|
135,503 |
|
|
|
15.64 |
|
|
|
3.54 |
|
|
$16.41 - $21.67 |
|
|
63,446 |
|
|
|
17.74 |
|
|
|
3.56 |
|
|
$21.68 - $22.28 |
|
|
60,700 |
|
|
|
22.28 |
|
|
|
2.67 |
|
|
|
|
|
1,024,529 |
|
|
$ |
10.54 |
|
|
|
5.58 |
|
As at December 31, 2016, the total unrecognized compensation expense related to stock options granted was $0.6 million, which is expected to be recognized into expense over a period of approximately 1.3 years.
The estimated grant date fair value of stock options vested during the years ended December 31, 2016, 2015 and 2014 was $1.0 million, $1.3 million and $1.9 million, respectively.
The aggregate intrinsic value of options exercised was calculated as the difference between the exercise price of the stock options and the fair value of the underlying common stock as of the date of exercise. The aggregate intrinsic value of options exercised for the years ended December 31, 2016, 2015 and 2014 was zero, $2,787 and $51,100, respectively. At December 31, 2016, the aggregate intrinsic value of the outstanding options was zero and the aggregate intrinsic value of the exercisable options was zero.
[d] Restricted Stock Unit Awards
We grant restricted stock unit awards that generally vest and are expensed over a four year period. We also grant restricted stock unit awards that vest in conjunction with certain performance conditions to certain executive officers and key employees. At each reporting date, we are required to evaluate whether achievement of the performance conditions is probable. Compensation expense is recorded over the appropriate service period based upon our assessment of accomplishing each performance provision. For the years ended December 31, 2016, 2015 and 2014, $0.7 million, $1.1 million and $2.2 million, respectively, of stock based compensation expense was recognized related to these awards.
The following table summarizes our restricted stock unit award activity during the years ended December 31, 2016, 2015 and 2014:
|
|
|
|
|
|
|
Weighted |
|
|
|
|
|
Number |
|
|
Average |
|
||
|
|
|
of |
|
|
Grant Date |
|
||
|
|
|
Shares |
|
|
Fair Value |
|
||
|
Balance, January 1, 2014 |
|
|
356,589 |
|
|
$ |
12.06 |
|
|
Granted |
|
|
814,800 |
|
|
|
6.48 |
|
|
Vested |
|
|
(199,887 |
) |
|
|
7.00 |
|
|
Forfeited or expired |
|
|
(291,301 |
) |
|
|
10.93 |
|
|
Balance, December 31, 2014 |
|
|
680,201 |
|
|
$ |
7.34 |
|
|
Granted |
|
|
249,775 |
|
|
|
1.92 |
|
|
Vested |
|
|
(269,401 |
) |
|
|
8.08 |
|
|
Forfeited or expired |
|
|
(19,816 |
) |
|
|
7.27 |
|
|
Balance, December 31, 2015 |
|
|
640,759 |
|
|
$ |
4.92 |
|
|
Granted |
|
|
— |
|
|
|
— |
|
|
Vested |
|
|
(217,296 |
) |
|
|
5.23 |
|
|
Forfeited or expired |
|
|
(170,242 |
) |
|
|
5.07 |
|
|
Balance, December 31, 2016 |
|
|
253,221 |
|
|
$ |
4.56 |
|
76
As of December 31, 2016, we had approximately $0.8 million in total unrecognized compensation expense related to our restricted stock unit awards which is to be recognized over a weighted-average period of approximately 1.4 years.
[e] Stock Warrants
The following is a summary of outstanding warrants to purchase common stock at December 31, 2016:
|
|
|
Total |
|
|
|
|
|
|
|
|
|
|
|
Outstanding |
|
|
Exercise |
|
|
|
||
|
|
|
and |
|
|
price per |
|
|
|
||
|
|
|
Exercisable |
|
|
Share |
|
|
Expiration Date |
||
|
(1) Series A Warrants issued in July 2014 financing |
|
|
2,779,933 |
|
|
|
4.00 |
|
|
July 2019 |
|
(2) Series B Warrants issued in July 2014 financing |
|
|
670,269 |
|
|
|
4.00 |
|
|
July 2019 |
|
(3) Series A-1 Warrants issued in April 2015 financing |
|
|
239,234 |
|
|
|
2.40 |
|
|
October 2020 |
No warrants were exercised for the year ended December 31, 2016. For the year ended December 31, 2015, all the Pre-Funded Series B warrants were exercised at a per unit price of $0.01, a total of 1,340,538 shares of common stock were issued for proceeds of $13,405. No Series A and Series B warrants from the July 2014 financing were exercised in 2015.
The Series A and Series B warrants issued in the July 2014 financing are classified as liabilities. The estimated fair value of warrants issued and classified as liabilities is reassessed at each reporting date using the Black-Scholes option pricing model. The Series A-1 Warrants issued in the April 2015 financing are classified as equity and are not reassessed for their fair value at the end of each reporting date. The following assumptions were used to value the warrants that are classified as liabilities on the following reporting dates:
|
|
|
As of |
|
|||||
|
|
|
December 31, |
|
|||||
|
Series A and Series B Warrant Valuation Assumptions |
|
2016 |
|
|
2015 |
|
||
|
Risk-free interest rates |
|
|
1.33 |
% |
|
|
1.42 |
% |
|
Expected dividend yield |
|
|
0 |
% |
|
|
0 |
% |
|
Expected life |
|
2.50 years |
|
|
3.50 years |
|
||
|
Expected volatility |
|
|
95 |
% |
|
|
77 |
% |
[f] 401(k) Plan
We maintain a 401(k) plan. Following the Arrangement, the Board of Directors of OncoGenex amended and restated the 401(k) plan whereas our securities are no longer offered as an investment option. This amendment prohibits the inclusion of our shares in the 401(k) plan, as well as any match of our shares to employee contributions.
[g] Loss per common share
The following table presents the computation of basic and diluted net loss attributable to common stockholders per share (in thousands, except per share and share amounts):
|
|
|
Years ended December 31, |
|
|||||||||
|
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|||
|
Numerator |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(20,129 |
) |
|
$ |
(16,801 |
) |
|
$ |
(26,240 |
) |
|
Denominator |
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of common shares outstanding |
|
|
29,949,432 |
|
|
|
26,147,344 |
|
|
|
18,098,799 |
|
|
Basic and diluted net loss per common share |
|
$ |
(0.67 |
) |
|
$ |
(0.64 |
) |
|
$ |
(1.45 |
) |
As of December 31, 2016, 2015 and 2014 a total of 5.3 million, 5.8 million and 7.0 million options, restricted stock units and warrants, respectively, have not been included in the calculation of potential common shares as their effect on diluted per share amounts would have been anti-dilutive.
77
11. RELATED PARTY TRANSACTIONS
In January 2016, Scott Cormack, our Chief Executive Officer, married Michelle Griffin, a consultant to us. For the twelve months ended December 31, 2016, we paid Ms. Griffin approximately $0.5 million for consulting services pursuant to a consulting agreement entered into in 2013 and amended thereafter. We also granted Ms. Griffin options to purchase 135,000 shares of common stock in 2016. In addition, pursuant to the consulting agreement with Ms. Griffin, as at December 31, 2016, we had an accrued termination liability of approximately $0.4 million.
12. COMMITMENTS AND CONTINGENCIES
The following table summarizes our contractual obligations as of December 31, 2016 (in thousands):
|
|
|
Total |
|
|
Less than 1 year |
|
|
1-3 years |
|
|
3-5 years |
|
|
More than 5 years |
|
|||||
|
Bothell office operating lease |
|
$ |
376 |
|
|
$ |
281 |
|
|
$ |
95 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Vancouver office operating lease |
|
$ |
68 |
|
|
$ |
68 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
UBC license maintenance fees |
|
$ |
26 |
|
|
$ |
4 |
|
|
$ |
9 |
|
|
$ |
9 |
|
|
$ |
4 |
|
|
Leased equipment |
|
$ |
22 |
|
|
$ |
19 |
|
|
$ |
3 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Total |
|
$ |
492 |
|
|
$ |
372 |
|
|
$ |
107 |
|
|
$ |
9 |
|
|
$ |
4 |
|
Teva Pharmaceutical Industries Ltd.
In December 2009, we, through our wholly-owned subsidiary, OncoGenex Technologies, entered into a Collaboration Agreement with Teva for the development and global commercialization of custirsen (and related compounds). In December 2014, we and Teva agreed to terminate the Collaboration Agreement upon entry into a Termination Agreement. In April 2015, we and Teva entered into the Termination Agreement, pursuant to which the Collaboration Agreement was terminated and we regained rights to custirsen. Pursuant to the Termination Agreement, Teva paid to us, as advanced reimbursement for certain continuing research and development activities related to custirsen, an amount equal to $27.0 million less approximately $3.8 million, which reduction represented a hold-back amount of $3.0 million and $0.8 million for certain third-party custirsen-related development expenses incurred by Teva between January 1, 2015 and the Closing Date. Pursuant to the Termination Agreement, we received a nominal amount from the remaining hold-back after deductions by Teva for certain costs incurred after the Closing Date. We do not expect to receive any additional amounts from Teva.
All licenses granted by us to Teva under the Collaboration Agreement were terminated as of the Closing Date.
In accordance with the Termination Agreement, Teva transferred certain third-party agreements for the ENSPIRIT study and custirsen development activities to us on the Closing Date. If any additional historical third-party agreements are discovered after the Closing Date and are used to conduct the ENSPIRIT study, then Teva will use commercially reasonable effort to assign such agreements to us and will be responsible for any costs invoiced under such agreements in excess of an aggregate of $0.1 million. We will be responsible for the initial $0.1 million of costs under such agreements.
Prior to the termination of the Collaboration Agreement, Teva made upfront payments in the aggregate amount of $50.0 million. Teva also acquired $10.0 million of our common stock at a premium under a separate Stock Purchase Agreement. We were required to contribute $30.0 million in direct and indirect costs towards the clinical development plan. We fulfilled our obligation to contribute $30.0 million towards the development of custirsen. Teva was required to and did fund all additional expenses under the clinical development plan through December 31, 2014, after which date we took over responsibility for future costs following termination of our Collaboration Agreement. We do not owe, to Teva, any development milestone payments or royalty payments on sales of custirsen, if any.
Ionis Pharmaceuticals Inc. and University of British Columbia
Custirsen
In November 2016, we provided the Notice of Discontinuance to Ionis and the Letter of Termination to UBC, notifying the parties that we have discontinued development of custirsen, resulting in termination of all licensing agreements related to custirsen. We believe that all financial obligations, other than continuing mutual indemnification obligations and our requirement to pay for out-of-pocket patent expenses incurred up to the date of termination and for abandoning the custirsen patents and patent applications, under all agreements with Ionis and UBC, including the Ionis settlement agreement, are no longer owed and no further payments are due.
Under the license agreements with Ionis and UBC, we were required to pay royalties to each of Ionis and UBC based on a percentage of net sales. We did not make any royalty payments to either Ionis or UBC in the nine months ended September 30, 2016. In addition,
78
pursuant to the terms of the agreements with Ionis, we were required to pay to Ionis up to 20% of all non-royalty revenue (defined to mean revenue not based on net sales of products) we receive from third parties.
In May and November 2015, we received communications from Ionis requesting payment of 30% of the $23.2 million paid by Teva under the Termination Agreement, as well as 30% of any amounts paid by Teva upon release of the $3.0 million holdback amount. In January 2016, Ionis filed a lawsuit and claimed that we were in breach of the license agreement for failing to pay Ionis a share of the advance reimbursement payment from Teva and other non-monetary consideration received from Teva in connection with the termination of the Collaboration Agreement. Ionis sought damages and a declaratory judgment that, based on our alleged breach, Ionis has the right to terminate the license agreement.
In August 2016, we and Ionis settled this lawsuit. Pursuant to the settlement, we paid to Ionis a $1.4 million upfront payment. In addition, under the settlement agreement, we were required to pay to Ionis additional success-based payments of up to an amount that does not exceed $5.0 million based on, (i) an additional 5% royalty on net sales of custirsen and (ii) 50% of any money we receive related to the sale, license or any other commercial transaction involving custirsen, subject to certain limitations. As a result of the Notice of Discontinuance, we believe that all financial obligations under the settlement agreement are no longer owed and no further payments are due.
Apatorsen and OGX-225
We are obligated to pay milestone payments of up to CAD $1.6 million and $7.75 million pursuant to license agreements with UBC and Ionis, respectively, upon the achievement of specified product development milestones related to apatorsen and OGX-225 and low to mid-single digit royalties on future product sales.
Unless otherwise terminated, the Ionis agreements for apatorsen will continue until the later of 10 years after the date of the first commercial product sale, or the expiration of the last to expire of any patents required to be licensed in order to use or sell the product, unless we discontinue apatorsen and Ionis does not elect to unilaterally continue development. The Ionis agreement for OGX-225 will continue into perpetuity unless we discontinue development of the product and Ionis does not elect to unilaterally continue development.
Lease Arrangements
We have an operating lease agreement for office space being used in Vancouver, Canada, which expires in September 2017. Pursuant to the operating lease agreement, we have the option to terminate the lease early without penalty at any time after January 1, 2017 so long as we provide three months prior written notice to the landlord.
The future minimum annual lease payments under the Vancouver lease is $68,000 in 2017.
In February 2015, we entered into an office lease with Grosvenor International (Atlantic Freeholds) Limited, or Landlord, pursuant to which we leased approximately 11,526 square feet located at 19820 North Creek Parkway, Bothell, Washington, 98011, commencing on February 15, 2015. The initial term of this lease will expire on April 30, 2018, with an option to extend the term for one approximately three-year period. Our monthly base rent for the premises will start at approximately $18,000 commencing on May 1, 2015 and will increase on an annual basis up to approximately $20,000. We received a construction allowance, for leasehold improvements that we made, of approximately $0.1 million. We will be responsible for 17% of taxes levied upon the building during each calendar year of the term. We delivered to the Landlord a letter of credit in the amount of $0.2 million, in accordance with the terms if the lease, which the Landlord may draw upon for base rent or other damages in the event of our default under this lease. In August 2015 we exercised our expansion option for an additional 2,245 square feet of office space, which commenced on August 1, 2015.
The remaining future minimum annual lease payments under the terminated Bothell lease are as follows (in thousands):
|
2017 |
|
|
281 |
|
|
2018 |
|
|
95 |
|
|
Total |
|
$ |
376 |
|
Consolidated rent and operating expense relating to both the Vancouver, Canada and Bothell, Washington offices for years ended December 31, 2016, 2015 and 2014 was $0.6 million, $0.9 million and $2.8 million, respectively.
79
In February 2015, we entered into a Lease Termination Agreement with BMR pursuant to which we and BMR agreed to terminate our lease, dated November 21, 2006, as amended, for the premises located at 1522 217th Place S.E. in Bothell, Washington, or Terminated Lease, effective March 1, 2015. Under the Lease Termination Agreement, we paid BMR a $2.0 million termination fee. BMR drew approximately $0.1 million on our letter of credit with respect to its payment of deferred state sales tax and terminated the remaining balance of $0.2 million. BMR returned to us the security deposit under the Terminated Lease, less amounts deducted in accordance with the terms of the Terminated Lease, of $0.5 million.
Pursuant to the Lease Termination Agreement, an additional termination fee of $1.3 million would have been payable to BMR if we had (i) met the primary endpoint for our phase 3 clinical trial for the treatment of second line metastatic castrate resistant prostate cancer, or CRPC, with custirsen, or the AFFINITY Trial, and if we had (ii) closed a transaction or transactions pursuant to which we received funding in an aggregate amount of at least $20.0 million. As at December 31, 2014 and subsequent annual and interim reporting periods up to June 30, 2016, we had assessed that the likelihood of meeting both contingent events was probable and as a result, recognized the $1.3 million in lease termination liability on our balance sheet as at the end of those reporting periods. In August 2016, final survival results of our AFFINITY trial did not meet the primary endpoint of a statistically significant improvement in overall survival in men with metastatic CRPC. As at September 30, 2016, we had re-assessed that the likelihood of meeting both contingent events is no longer possible due to not achieving the primary endpoint on our AFFINITY trial. As a result, we have reversed the $1.3 million in lease termination liability on our balance sheet as at September 30, 2016 and recognized a recovery on our statement of loss.
Change in Control and Severance Agreements
Our officers and certain employees have agreements which provide for payouts in the event that we consummate a change in control. In addition, our officers and certain employees are also entitled to full vesting of their outstanding equity awards. These agreements also provide for customary severance compensation. As of December 31, 2016 and 2015 we did not consummate any change in control transaction. See also Note 13.
Guarantees and Indemnifications
We indemnify our officers, directors and certain consultants for certain events or occurrences, subject to certain limits, while the officer or director is or was serving at its request in such capacity. The term of the indemnification period is equal to the officer’s or director’s lifetime.
The maximum amount of potential future indemnification is unlimited; however, we have obtained director and officer insurance that limits our exposure and may enable us to recover a portion of any future amounts paid. We believe that the fair value of these indemnification obligations is minimal. Accordingly, we have not recognized any liabilities relating to these obligations as of December 31, 2016.
We have certain agreements with certain organizations with which it does business that contain indemnification provisions pursuant to which it typically agrees to indemnify the party against certain types of third-party claims. We accrue for known indemnification issues when a loss is probable and can be reasonably estimated. There were no accruals for or expenses related to indemnification issues for any period presented.
Material Changes in Financial Condition
|
|
|
December 31, |
|
|||||
|
(in thousands) |
|
2016 |
|
|
2015 |
|
||
|
Total Assets |
|
$ |
27,470 |
|
|
$ |
58,209 |
|
|
Total Liabilities |
|
|
8,504 |
|
|
|
20,769 |
|
|
Total Equity |
|
|
18,966 |
|
|
|
37,440 |
|
The decrease in assets at December 31, 2016 compared with December 31, 2015 was due to a decrease in cash and cash equivalents as these assets have been used to fund operations and a decrease in prepaid assets related to the drawdown of our escrow payments to our clinical research organization vendors. The decrease in liabilities at December 31, 2016 compared with December 31, 2015 was due to a decrease in clinical trial accruals associated with patient treatment costs in the AFFINITY trial, ENSPIRIT trial and our investigator sponsored trials evaluating apatorsen, lower deferred revenue as these amounts were recognized into collaboration revenue on a dollar for dollar basis as costs were incurred as part of the continuing research and development activities related to custirsen, the reversal of the lease termination liability and decrease in accrued compensation liabilities. This was partially offset by higher accrued liabilities other as a result of the severance associated with the restructurings announced in fiscal 2016.
80
Restructure
In February 2016, we committed to a plan to reduce operating expenses, which included a workforce reduction of 11 employees, representing approximately 27% of our employees prior to the reduction. We incurred approximately $0.4 million in expenses as a result of the workforce reduction, substantially all of which were severance costs.
In October 2016, we committed to a restructuring of an additional portion of our workforce in order to preserve our resources as we determine future strategic plans. As part of this restructuring, we eliminated 14 positions, representing approximately 48% of our workforce. We expect the restructuring to be substantially complete in the first quarter of 2017. As of December 31, 2016, we incurred approximately $1.1 million in restructuring costs, substantially all of which related to severance costs.
In November 2016, we committed to a further reduction in our workforce. We eliminated five positions and incurred approximately $0.7 million in expenses as a result of the workforce reduction, substantially all of which were severance costs.
|
|
|
Total |
|
|
Amounts |
|
|
Accrued at |
|
|||
|
|
|
estimated |
|
|
settled |
|
|
December 31, |
|
|||
|
|
|
costs |
|
|
to date |
|
|
2016 |
|
|||
|
Restructuring Costs |
|
$ |
2,206 |
|
|
$ |
(708 |
) |
|
$ |
1,498 |
|
14. QUARTERLY FINANCIAL INFORMATION (UNAUDITED)
The following table summarizes the unaudited statements of operations for each quarter of 2016 and 2015 (in thousands, except per share amounts):
|
|
|
March 31 |
|
|
June 30 |
|
|
September 30 |
|
|
December 31 |
|
||||
|
2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Collaboration revenue |
|
$ |
2,940 |
|
|
$ |
2,122 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Research and development |
|
|
4,642 |
|
|
|
4,662 |
|
|
|
3,782 |
|
|
|
1,702 |
|
|
General and administrative |
|
|
2,299 |
|
|
|
2,475 |
|
|
|
1,864 |
|
|
|
2,295 |
|
|
Restructuring costs (recovery) |
|
|
431 |
|
|
|
(8 |
) |
|
|
(31 |
) |
|
|
1,814 |
|
|
Recovery of lease termination loss |
|
|
— |
|
|
|
— |
|
|
|
(1,250 |
) |
|
|
— |
|
|
Litigation settlement |
|
|
— |
|
|
|
1,375 |
|
|
|
— |
|
|
|
— |
|
|
Asset impairment charge |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
202 |
|
|
Total expenses |
|
|
7,372 |
|
|
|
8,504 |
|
|
|
4,365 |
|
|
|
6,013 |
|
|
Other income |
|
|
725 |
|
|
|
(507 |
) |
|
|
675 |
|
|
|
170 |
|
|
Net loss |
|
|
(3,707 |
) |
|
|
(6,889 |
) |
|
|
(3,690 |
) |
|
|
(5,843 |
) |
|
Basic and diluted net loss per share |
|
$ |
(0.12 |
) |
|
$ |
(0.23 |
) |
|
$ |
(0.12 |
) |
|
$ |
(0.19 |
) |
|
2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Collaboration revenue |
|
$ |
1,374 |
|
|
$ |
4,025 |
|
|
$ |
6,737 |
|
|
$ |
6,024 |
|
|
Research and development |
|
|
3,673 |
|
|
|
6,545 |
|
|
|
8,303 |
|
|
|
6,587 |
|
|
General and administrative |
|
|
2,698 |
|
|
|
3,067 |
|
|
|
3,125 |
|
|
|
2,915 |
|
|
Total expenses |
|
|
6,371 |
|
|
|
9,612 |
|
|
|
11,428 |
|
|
|
9,502 |
|
|
Other income |
|
|
480 |
|
|
|
(423 |
) |
|
|
141 |
|
|
|
1,756 |
|
|
Net loss |
|
|
(4,517 |
) |
|
|
(6,010 |
) |
|
|
(4,550 |
) |
|
|
(1,722 |
) |
|
Basic and diluted net loss per share |
|
$ |
0.20 |
|
|
$ |
0.26 |
|
|
$ |
0.16 |
|
|
$ |
0.06 |
|
15. SUBSEQUENT EVENTS
On January 5, 2017, we and Achieve entered into the Merger Agreement, pursuant to which Ash Acquisition Sub, Inc., a Delaware corporation and a wholly owned subsidiary of ours will merge with and into Achieve, or the First Merger, with Achieve becoming a wholly owned subsidiary of ours and the surviving company of the First Merger, or the Initial Surviving Corporation. Promptly following the First Merger, the Initial Surviving Corporation will merge with and into Ash Acquisition Sub 2, Inc., or Merger Sub 2, a Delaware corporation and a wholly owned subsidiary of ours, with Merger Sub 2 continuing as the surviving entity as a direct wholly owned subsidiary of ours. The two mergers taken together, are intended to qualify as a “reorganization” within the meaning of Section 368(a)(2)(D) of the Internal Revenue Code of 1986, as amended. The surviving company is expected to be renamed Achieve Life Sciences, Inc. and is referred to herein as the “combined company.” The Merger is expected to close mid-2017.
81
Subject to the terms and conditions of the Merger Agreement, at the closing of the First Merger, each outstanding share of Achieve common stock will be converted into the right to receive approximately 4,242.8904 shares of our common stock, subject to adjustment as provided in the Merger Agreement based on increases or decreases in Achieve’s fully-diluted capitalization, as well as the payment of cash in lieu of fractional shares. Immediately following the effective time of the merger, our equityholders are expected to own approximately 25% of the outstanding capital stock of the combined company on a fully diluted basis, and the Achieve stockholders are expected to own approximately 75% of the outstanding capital stock of the combined company on a fully diluted basis.
Consummation of the merger is subject to certain closing conditions, including, among other things, approval by the stockholders of us and Achieve. The Merger Agreement contains certain termination rights for both us and Achieve, and further provides that, upon termination of the Merger Agreement under specified circumstances, either party may be required to pay the other party a termination fee of $0.5 million. In addition, the Merger Agreement provides that if either party breaches certain covenants regarding alternative transactions to those contemplated by the Merger Agreement, the breaching party may be required to pay the other party a termination fee of $1.0 million. In connection with certain terminations of the Merger Agreement, either party may be required to pay the other party’s third party expenses up to $0.5 million.
At the effective time of the First Merger, our Board of Directors is expected to consist of seven members, three of whom will be designated by us and four of whom will be designated by Achieve. We are expected to designate Scott Cormack, Stewart Parker and Martin Mattingly. Achieve is expected to designate Richard Stewart, Anthony Clark and two other independent directors that have yet to be determined. Additionally, at the effective time of the First Merger, Rick Stewart, the current Chairman of Achieve, is expected to be the Chairman and Chief Executive Officer of the combined company; Anthony Clarke, the current Chief Scientific Officer of Achieve, is expected to be the Chief Scientific Officer of the combined company; and John Bencich, our Chief Financial Officer and Cindy Jacobs, our Chief Medical Officer, are expected to continue to serve the combined company in their respective roles.
In accordance with the terms of the Merger Agreement, (i) certain of our officers and directors, who collectively hold approximately 1.2 percent of the outstanding shares of our capital stock as of the close of business on January 4, 2017, have each entered into a support agreement with Achieve, or the OncoGenex Support Agreements, and (ii) certain officers, directors and stockholders of Achieve, who collectively hold approximately 78 percent of the outstanding shares of Achieve capital stock as of the close of business on January 4, 2017, have each entered into a support agreement with us, or the Achieve Support Agreements, and together with the OncoGenex Support Agreements, the Support Agreements. The Support Agreements include covenants as to the voting of such shares in favor of approving the transactions contemplated by the Merger Agreement and against actions that could adversely affect the consummation of the Merger.
The Support Agreements will terminate upon the earlier of the consummation of the First Merger or the termination of the Merger Agreement by its terms.
Concurrently and in connection with the execution of the Merger Agreement, (i) certain of our officers and directors, who collectively hold approximately 1.2 percent of the outstanding shares of our capital stock as of the close of business on January 4, 2017 and (ii) certain officers, directors and stockholders of Achieve, who collectively hold approximately 78 percent of the outstanding shares of Achieve capital stock as of the close of business on January 4, 2017, have each entered into lock-up agreements with us, pursuant to which, subject to certain exceptions, each stockholder will be subject to a 180-day, or the Lock-Up Period, lock-up on the sale of shares of our capital stock, which Lock-Up Period shall begin upon the consummation of the First Merger.
We expect to issue contingent value rights, or each, a CVR and collectively, the CVRs, to our existing stockholders prior to the completion of the First Merger. One CVR will be issued for each share of our common stock outstanding as of the record date for such issuance. Each CVR will be a non-transferable right to potentially receive certain cash, equity or other consideration received by the combined company in the event the combined company receives any such consideration during the five-year period after consummation of the First Merger as a result of the achievement of certain clinical milestones, regulatory milestones, sales-based milestones and/or up-front payment milestones relating to our product candidate apatorsen, or the Milestones, upon the terms and subject to the conditions set forth in a contingent value rights agreement to be entered into between us, Achieve and an as of yet unidentified third party, as rights agent, or the CVR Agreement. The aggregate consideration to be distributed to the holders of the CVRs, if any, will be equal to 80% of the consideration received by the combined company as a result of the achievement of the Milestones less certain agreed to offsets, as determined pursuant to the CVR Agreement. Under the CVR Agreement, for a period of six months beginning in February 2017, we will use certain defined efforts to enter into an agreement with a third party regarding the development and/or commercialization of apatorsen. At the expiration of this six-month period, if a third party has not entered into a term sheet for the development or commercialization of apatorsen, the combined company will no longer be contractually required to pursue an agreement regarding apatorsen and no consideration will be payable to the holders of CVRs.
We also entered into a letter agreement with Achieve, whereby we would pay, on behalf of Achieve, for transactions costs associated with the merger. In the event that the Merger Agreement is terminated and as a result of such termination we are required to pay to
82
Achieve one or more termination fees, the total amount of termination fees we would owe is reduced by the amount of the transaction costs we would have paid on behalf of Achieve.
In January 2017, we discontinued further development of OGX-225. We provided a notice of discontinuance to Ionis, notifying them that we have discontinued development of OGX-225 resulting in termination of the license agreement related to this product candidate. We intend to also terminate the UBC license agreement related to OGX-225 provided that Ionis does not exercise its reversion rights within 90 days of the notice of discontinuance.
83
None.
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that material information required to be disclosed in our periodic reports filed or submitted under the Exchange Act, is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Our disclosure controls and procedures are also designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure.
We carried out an evaluation, under the supervision and with the participation of our management, including the principal executive officer and the principal financial officer, of the effectiveness of the design and operation of the disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act. Based on that evaluation, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures were effective as of the end of the period covered by this Annual Report on Form 10-K.
Changes in Internal Control Over Financial Reporting
We have not made any changes to our internal control over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act) during the quarter ended December 31, 2016 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting, as defined in Rule 13a-15(f) under the Exchange Act. Our internal control over financial reporting is a process designed under the supervision of our principal executive and principal financial officers to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our financial statements for external reporting purposes in accordance with U.S. generally accepted accounting principles.
As of December 31, 2016, management assessed the effectiveness of our internal control over financial reporting based on the framework established in “Internal Control—Integrated Framework” issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) (2013 Framework). Based on this evaluation, management has determined that our internal control over financial reporting was effective as of December 31, 2016.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The effectiveness of our internal control over financial reporting as of December 31, 2016 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report which is included above.
Not applicable.
84
Executive Officers
The following table provides information regarding our current executive officers as of February 14, 2017.
|
Name |
Age |
Position With the Company |
|
|
|
|
|
Scott Cormack, |
51 |
President, Chief Executive Officer, Treasurer, Secretary and Director |
|
|
|
|
|
Cindy Jacobs, |
59 |
Executive Vice President and Chief Medical Officer |
|
|
|
|
|
John Bencich, |
39 |
Vice President and Chief Financial Officer |
Following are the biographies of the foregoing persons, except the biography of Mr. Cormack, which is located below under the heading “Board of Directors.”
Cindy Jacobs, Ph.D., M.D., 59 has served as our Executive Vice President and Chief Medical Officer since August 2008, and had been Executive Vice President and Chief Medical Officer of OncoGenex Technologies Inc. from September 2005 to August 2008. From 1999 to July 2005, Dr. Jacobs served as Chief Medical Officer and Senior Vice President, Clinical Development of Corixa Corporation. Prior to 1999, Dr. Jacobs held Vice President, Clinical Research positions at two other biopharmaceutical companies. Dr. Jacobs received her Ph.D. degree in Veterinary Pathology/Microbiology from Washington State University and an M.D. degree from the University of Washington Medical School.
John Bencich, 39, has served as our Vice President and Chief Financial Officer since August 2014. Mr. Bencich joined us from Integrated Diagnostics, Inc., a molecular diagnostics company, where he served as Chief Financial Officer from September 2012 to August 2014. Prior to joining Integrated Diagnostics, he served as Chief Financial Officer of Allozyne, Inc. since July, 2011. Mr. Bencich was an independent consultant from November 2010 until he joined Allozyne. He served as the Vice President, Chief Financial Officer and Treasurer of Trubion Pharmaceuticals, Inc., a biotechnology company, from November 2009 until its acquisition by Emergent BioSolutions Inc. in October 2010. Mr. Bencich served as Trubion’s Senior Director of Finance and Accounting from May 2007 through November 2009. From September 2004 to April 2007, Mr. Bencich was an employee of Onyx Software Corporation, a software company, where he last served as Director of Finance and Corporate Controller. From 1999 to 2004, Mr. Bencich was an employee of Ernst & Young LLP, an international professional services firm, where he last served as a manager. Mr. Bencich received a B.A. in Accountancy from the University of San Diego and an M.B.A. from Seattle University. Mr. Bencich received his Certified Public Accountant Certification from the State of Washington and currently holds an active license.
85
Directors are elected at each annual stockholders meeting to hold office until the next annual meeting or until their successors are elected and have qualified. Currently, there are six members of the Board of Directors. The following table sets forth information with respect to our current directors. The ages of such persons are shown as of February 14, 2017.
|
Name and Municipality of Residence |
Age |
Position |
Director Since |
|
|
|
|
|
|
Scott Cormack |
51 |
Director, President and Chief Executive Officer |
2008 |
|
|
|
|
|
|
Neil Clendeninn |
67 |
Director, Chairperson of the Compensation Committee and Member of the Nominating and Governance Committee |
2008 |
|
|
|
|
|
|
Jack Goldstein |
69 |
Chairperson of the Board of Directors, Member of the Compensation Committee, Member of the Nominating and Governance Committee and Member of the Audit Committee |
2010 |
|
|
|
|
|
|
Martin Mattingly |
60 |
Director, Member of the Compensation Committee and Member of the Audit Committee |
2010 |
|
|
|
|
|
|
Stewart Parker |
61 |
Director, Chairperson of the Nominating and Governance Committee and Member of the Audit Committee |
2010 |
|
|
|
|
|
|
David Smith Alamo, California |
57 |
Director, Chairperson of the Audit Committee and Member of the Nominating and Governance Committee |
2010 |
Following are the biographies of the foregoing persons.
Scott Cormack, 51, has been our President, Chief Executive Officer and a director since August 2008. He was a co-founder of OncoGenex Technologies Inc., which is our wholly owned subsidiary, and has been its President since May 2000, its Chief Executive Officer since February 2002 and a member of its Board of Directors since May 2000. Mr. Cormack currently serves on the Board of Directors of the Prostate Centre’s Translation Research Initiative for Accelerated Discovery and Development and the Board of Directors of the Prostate Centre at Vancouver General Hospital. Mr. Cormack served as interim President, Chief Executive Officer and Chairman of the Board of Directors of Salpep Biotechnology Inc., an asthma and inflammation biotechnology company, from 2000 to 2001 and on the Board of Directors of Aurinia Pharmaceuticals from 2012 to 2014. From 1998 to 2001, Mr. Cormack served as Vice President of Milestone Medica Corporation, a seed venture capital firm investing in life sciences opportunities. Mr. Cormack holds a B.S. degree from the University of Alberta. The determination was made that Mr. Cormack should serve on our Board of Directors due to our belief that it is of importance that the Board of Directors have the benefit of management’s perspective and, in particular, that of the Chief Executive Officer.
Neil Clendeninn, M.D., Ph.D., 67, has served as a director since August 2008. Additionally, he has served as a member of OncoGenex Technologies Inc.’s Board of Directors since September 2004. Dr. Clendeninn served as Senior Vice President and Chief Medical Officer of Heron Therapeutics from October 2015 to September 2016. Additionally, Dr. Clendeninn is currently a practicing physician and serves as Program Director for Palliative Medicine Partners: Complex Illness Coordination, a program of Kauai Hospice in Kauai, Hawaii. Dr. Clendeninn served as Corporate Vice President, Head of Clinical Affairs of Agouron Pharmaceuticals, Inc., a biopharmaceutical company and a subsidiary of Pfizer Inc., a pharmaceutical company, from 1993 until his retirement in 2001. Dr. Clendeninn holds a B.A. degree in biology/chemistry from Wesleyan University, and a Ph.D. degree in microbiology/pharmacology and an M.D. degree from New York University. The determination was made that Dr. Clendeninn should serve on our Board of Directors due to his training and experience as a medical oncologist and his executive-level experience in public development-stage oncology-focused companies.
Jack Goldstein, Ph.D., 69, has served as our Chairman of the Board of Directors since March 2010. Dr. Goldstein was President and Chief Operating Officer of Chiron Corporation, a biotechnology company, from November 2004 until its acquisition by Novartis AG in April 2006, prior to which he served as Vice President and President, Chiron Blood Testing Division beginning in 2002. From 2000 to 2002, Dr. Goldstein was General Partner at Windamere Venture Partners, L.L.C., a venture capital fund. From 1997 to 2001, he served as President and Chief Executive Officer of Applied Imaging Corporation, a supplier of instrument systems for prenatal and cancer genetics, where he also served as Chairman of the Board of Directors from 1999 to 2002. Dr. Goldstein currently serves on the Board of Directors of Accuray Incorporated and Counsyl, Inc. and has served as a director of Orasure Technologies Inc. from May 2006 to May 2011, Illumina, Inc. from June 2006 to May 2010, and Immucor, Inc. from December 2007 to February 2009.
86
Dr. Goldstein holds a B.A. degree in biology from Rider University, and a M.S. degree in immunology and a Ph.D. degree in microbiology from St. John’s University. The determination was made that Dr. Goldstein should serve on our Board of Directors as a result of his extensive experience as a senior executive and as a chair of the board of directors of both publicly held and privately held biotechnology or pharmaceutical companies.
Martin Mattingly, Pharm.D., 60, has served as a director since June 2010. Since August 2012, Dr. Mattingly has served as a member of Tech Coast Angels, an angel investor group, and since December 2014 has served as a director of TRACON Pharmaceuticals, Inc. Previously, Dr. Mattingly served as the Chief Executive Officer of Trimeris, Inc., a biopharmaceutical company, from November 2007 until its merger with Synageva in November 2011. He also served on the Board of Directors of Trimeris, Inc. from November 2007 until November 2011. From 2005 to 2007, Dr. Mattingly was employed at Ambrx, Inc., a biopharmaceutical company, where he served as President and Chief Executive Officer. From 2003 to 2005, Dr. Mattingly served as Executive Vice President and Chief Operating Officer of CancerVax Corporation, a biotechnology company. From 1996 to 2003, he provided senior leadership in various management positions at Agouron Pharmaceuticals, Inc. and Pfizer, Inc., including serving as General Manager of the Agouron HIV division, Vice President, Product Development Group at Pfizer and Vice President, Global Marketing Planning at Pfizer. Dr. Mattingly holds a Pharm.D. degree from the University of Kentucky. The determination was made that Dr. Mattingly should serve on the Board of Directors as a result of his executive leadership experience in late-stage clinical development, public company expertise, and commercialization and business development experience with pharmaceuticals and biologics.
Stewart Parker, 61, has served as a director since March 2010. Ms. Parker served as the Chief Executive Officer of the Infectious Disease Research Institute, or IDRI, a nonprofit research organization focused on the development of products for the diagnosis, prevention, and treatment of neglected diseases from March 2011 to January 2014. Prior to IDRI, Ms. Parker managed the formation of Targeted Genetics Corporation, a biotechnology company, as a wholly owned subsidiary of Immunex Corporation, a biotechnology company, and served as its President and Chief Executive Officer and as a director from its spinout from Immunex Corporation in 1992 to November 2008. She served in various capacities at Immunex Corporation from August 1981 through December 1991, most recently as Vice President, Corporate Development. Ms. Parker currently serves on the Board of Directors of Sangamo BioSciences since June, 2014. She served on the Board of Directors and the executive committee of BIO, the primary trade organization for the biotechnology industry. Ms. Parker has also served as a director of Targeted Genetics Corporation from 1992 to November 2008 and Neose Technologies, Inc. from May 2005 to January 2009. Ms. Parker received her B.A. and M.B.A. degrees from the University of Washington. The determination was made that Ms. Parker should serve on our Board of Directors due to her executive leadership experience in development-stage clinical development, public company expertise, and business development experience for pharmaceuticals and biologics.
David Smith, 57, has served as a director since August 2010. Since June 2012 Mr. Smith has served as Chief Operating Officer of IntegenX, Inc., a molecular diagnostics company. From December 2006 to July 2011, Mr. Smith was the Executive Vice President and Chief Financial Officer of Thoratec Corporation, a medical device company. Mr. Smith served as the Vice President and Chief Financial Officer of Chiron Corporation from April 2003 to April 2006, as its Vice President, Finance from February 2002 to April 2003 and as its Vice President and Principal Accounting Officer from February 1999 to February 2002. Mr. Smith earned a B.A degree in economics and history from Willamette University and a M.B.A degree in finance from Golden Gate University. Mr. Smith also serves as a director of Codexis, Inc. The determination was made that Mr. Smith should serve on our Board of Directors due to his financial expertise and extensive experience as a senior executive at publicly held biotechnology companies.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires that our executive officers and directors, and persons who own more than 10% of a registered class of our equity securities, file reports of ownership and changes in ownership with the SEC. Executive officers, directors and greater than 10% stockholders are required by SEC rules to furnish us with copies of all forms they file. Based solely on our review of the copies of such forms we received and written representations from certain reporting persons, we believe that all of our executive officers, directors and 10% stockholders timely filed all reports required to be filed under Section 16(a) during fiscal year 2015.
We believe that sound corporate governance policies are essential to earning and retaining the trust of investors. We are committed to maintaining the highest standards of integrity. We have adopted a Code of Business Conduct and Ethics that is applicable to our principal executive officer, our principal financial officer and our principal accounting officer, as well as to all of our other employees and directors, and have posted such code on our website at http://ir.oncogenex.com/governance.cfm.
87
Stockholder Nominations and Recommendations for Director Candidates
We have not made any material changes to the procedures by which our stockholders may recommend nominees to our Board of Directors since we last disclosed the procedures by which stockholders may nominate director candidates under the caption “Board of Directors—Director Nomination Process” in our proxy statement for our 2016 annual meeting of stockholders filed with the SEC on April 21, 2016.
Audit Committee and Audit Committee Financial Expert
The Audit Committee has been established in accordance with Section 3(a)(58)(A) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and is currently comprised of David Smith (Chairperson), Jack Goldstein, Martin Mattingly and Stewart Parker, each of whom the Board of Directors has determined satisfies the applicable SEC and NASDAQ independence requirements for audit committee members. The Board of Directors has also determined that Mr. Smith is an “audit committee financial expert,” as defined by the applicable rules of the SEC
Executive Compensation Discussion and Analysis
Our executive compensation program is designed to:
|
|
• |
attract and retain the most talented and dedicated executives possible; |
|
|
• |
align our executive officers’ incentives with stockholder value creation; |
|
|
• |
correlate annual and long-term cash and stock incentives to achievement of measurable strategic performance objectives; and |
|
|
• |
increase the percentage of executive compensation that is performance-based, and therefore at-risk, as an executive’s experience, unique expertise and criticality of role increases. |
To achieve these objectives we have established compensation programs that tie a substantial portion of each executive’s overall compensation to key strategic operational and financial goals such as the development of our product candidates, the establishment and maintenance of key strategic relationships, and the identification and advancement of additional product candidates. The Compensation Committee’s approach emphasizes the setting of compensation at levels the committee believes are competitive with executives in other companies of similar size and stage of development operating in the biotechnology industry while taking into account our relative performance and our own strategic goals. Our annual cash incentives and a portion of our longer term incentives, such as our performance-based equity awards, are tied to our achievement of corporate operating goals. We believe that successful execution against goals is the best way to enhance long-term stockholder value. Overall, our pay programs attempt to balance cash and equity to reward both short- and long-term performance.
Compensation Determination Process and the Role of Executive Officers in Compensation Decisions
The compensation review process is conducted in January in order to facilitate the comparison of corporate objectives against our full year performance. The Chief Executive Officer provides a presentation regarding our current compensation philosophies and programs to the Compensation Committee with the remaining members of the Board of Directors invited to attend. Typically, the Chief Executive Officer produces an executive compensation review for each Named Executive Officer, excluding the Chief Executive Officer, which includes recommendations for:
|
|
• |
base salary for the upcoming year; |
|
|
• |
year-end cash incentive award, if any, under the terms of our discretionary short-term incentive awards program, or STIP, based on the achievement of corporate objectives; and |
|
|
• |
annual equity awards of stock options and restricted stock units, or RSUs. |
The Chief Executive Officer may also recommend other changes to an executive’s compensation package, such as changes in the executive’s eligibility for cash incentives. The Compensation Committee evaluates and, if determined appropriate or advisable, revises the Chief Executive Officer’s recommendations and forwards its own recommendations to the Board of Directors, which may in turn suggest further revisions.
The Compensation Committee also meets in executive sessions without the Chief Executive Officer present to discuss the Chief Executive Officer’s compensation, including base salary, year-end cash incentive award and annual stock option grant, and to make recommendations regarding such compensation to the Board of Directors. The Board of Directors considers the Compensation
88
Committee’s recommendations with respect to the Chief Executive Officer in executive session and provides feedback to the Compensation Committee. With the exception of executive sessions of the Compensation Committee and the Board of Directors to review, recommend and approve the Chief Executive Officer’s compensation, the Chief Executive Officer is generally present at all deliberations of the Compensation Committee and the Board of Directors related to executive compensation.
During the first meeting following the completion of the fiscal year, the Chief Executive Officer recommends to the Compensation Committee the corporate objectives to be adopted under the terms of the STIP for the upcoming year. The Compensation Committee evaluates and may revise the Chief Executive Officer’s recommendations and forwards its own recommendations to the Board of Directors, which may in turn suggest further revisions.
The Compensation Committee makes final determinations with respect to the award of cash incentives under the STIP and all annual equity awards. The Board of Directors, after reviewing the recommendations of the Compensation Committee, makes final determinations with respect to the cash incentives to the executive officers and the Chief Executive Officer, as well as the corporate objectives under the STIP. From time to time at the request of the Compensation Committee, members of our executive management team, including representatives from finance, legal and human resources, may provide information to the Compensation Committee and attend all or a portion of certain of the committee’s meetings.
Benchmarking of Executive Compensation
We participate annually in Radford’s Compensation Survey and in return we receive the Compensation Survey results. Additionally, every year or at the direction of the Compensation Committee, our management reviews peer group data compiled by Radford to determine whether total direct compensation and each component of the compensation package are approximately equal to the targeted 50th percentile for executive officer compensation of our peer group. The peer group companies are amended from time to time at the discretion of the Board of Directors and based on Radford recommendations. In 2015, our Compensation Committee engaged Radford to review compensation levels and executive agreements of our executive officers and to provide a report summarizing relevant benchmark data and making recommendations as to executive compensation levels. Radford’s review included benchmarking the base salary, target total cash (base salary plus target cash incentives) and long-term incentives of our executives with industry-appropriate peers based on the following characteristics:
|
|
• |
pre-commercial biotechnology/biopharmaceutical companies at a similar stage of drug development (Phase II to Phase III) |
|
|
• |
companies located in biotechnology hub markets (Seattle, San Francisco, San Diego and Boston) to reflect the recruiting market for executive talent; |
|
|
• |
companies with market values |
|
|
• |
companies with |
In addition, Radford also examined research and development spending, cash on-hand and total shareholder return over one and three years as additional metrics to help determine appropriate peer companies.
The peer group recommended by Radford in 2015 and approved by the Compensation Committee and the Board of Directors for the evaluation of compensation levels for the 2016 fiscal year, after a comprehensive review, analysis and discussion regarding Radford’s recommendations, is comprised of the companies set forth below.
|
Actinium Pharmaceuticals, Inc. |
MediciNova, Inc. |
|
Anthera Pharmaceuticals, Inc. |
MEI Pharma, Inc. |
|
ArQule Inc. |
Onconova Therapeutics, Inc. |
|
Aveo Pharmaceuticals, Inc. |
Oncothyreon Inc. |
|
BIND Therapeutics, Inc. |
OXiGENE, Inc. |
|
Celator Pharmaceuticals, Inc. |
Rexahn Pharmaceuticals, Inc. |
|
Celsion Corporation |
|
|
Conatus Pharmaceuticals Inc. |
Sunesis Pharmaceuticals, Inc. |
|
Cyclacel Pharmaceuticals, Inc. |
Targacept, Inc. |
|
|
TetraLogic Pharmaceuticals Corporation |
|
GTx, Inc. |
Threshold Pharmaceuticals, Inc. |
|
Kalobios Pharmaceuticals, Inc. |
Vical Incorporated |
89
Radford reports directly to our Compensation Committee and does not provide any services to us other than the services provided to the Compensation Committee and the preparation and delivery of their compensation survey, for which we pay a nominal fee. Our Compensation Committee believes that Radford does not have any conflicts of interest in advising the Compensation Committee under applicable SEC or NASDAQ rules.
Benchmarking in the Context of Our Other Executive Compensation Principles
In establishing executive compensation, the Compensation Committee focuses on a range around the 50th percentile of peer group benchmarking for each of base salary, target total cash (base salary plus target cash incentives) and long-term incentives for each similarly situated executive, which the Compensation Committee believes provides the tools to allow a company of our size to attract, compete for and retain the type of executives necessary for us to achieve our goals but conserve our cash and equity as much as possible.
The Compensation Committee realizes, however, that using a peer benchmark is neither the only means for gathering and validating market data nor the only criteria for establishing executive pay. In instances where an executive officer is uniquely critical to our success, the Compensation Committee may provide compensation in excess of the benchmark. Upward or downward variations for base salary and long-term incentives may also occur as a result of the individual’s experience level, the balance of the individual’s different elements of compensation, market factors and other strategic considerations, which we refer to below as compensation factors. Additional market surveys, such as the Radford Global Life Sciences Survey, which reports compensation practices of a broad range of life science companies, are also utilized in determining market competitive compensation. The Compensation Committee believes that, given the competitiveness of our industry and our company culture, our base compensation, cash incentives and equity programs are flexible, reward the achievement of clearly defined corporate goals and are generally sufficient to retain our existing executive officers and to hire new executive officers when necessary.
Elements of Executive Compensation
We have designed and implemented compensation policies that have historically allowed us to recruit both in our geographic areas of operation, which are Seattle, Washington and Vancouver, British Columbia, and other areas. We seek to implement and utilize compensation policies that balance fixed and variable pay costs for a long-term, sustainable approach to talent acquisition and retention. Our executive compensation consists of base salary, cash incentives and stock option and RSU grants, each of which is discussed in detail below.
Base Salary
We provide an annual salary based on comparable market data for level of responsibility, expertise, skills, knowledge, experience, our unique organizational requirements and desire to maintain internal equity. When establishing executive compensation for 2016, the Compensation Committee focused on the respective peer group market survey as well as the Radford Global Life Sciences Survey for base salaries and incentive compensation. The executive base salary program targets a range around the 50th percentile of salaries for executives with the requisite skills in similar positions with similar responsibilities at comparable companies. In reviewing base salaries annually, the Compensation Committee also considers the role, overall value and contribution each executive makes to the achievement of our objectives. Executives may be compensated below or above that range based on the compensation factors described above. The Compensation Committee reviews base salaries in the first quarter of each year and may make adjustments from time to time to realign salaries with market levels after taking into account the compensation factors.
Cash Incentives
The STIP provides an annual opportunity for our Named Executive Officers to receive a discretionary cash bonus (stated as a percentage of each officer’s salary) based on performance related to corporate objectives established by the Board of Directors. The STIP, when combined with each executive’s base salary, is designed to provide total target cash compensation within a range around the 50th percentile of our peer group, subject to adjustment for the compensation factors described above. For any given year, these objectives may relate to operational, strategic or financial factors such as developing our product candidates, establishing and maintaining key strategic relationships, raising or maintaining certain levels of capital, improving our results of operations or increasing the price per share of our common stock. The Compensation Committee alone determines achievement level of corporate objectives as it relates to incentive compensation for executive officers. If corporate objectives are not achieved at a 100% level, the Compensation Committee may determine that the corporate objectives were not achieved or, in its sole discretion, may determine that such objectives were partially achieved. The Compensation Committee may award bonuses based on the foregoing determinations or, after considering market conditions, our financial position or other factors, the Compensation Committee may, in its sole discretion, determine not to award any bonuses or to award bonuses at less than maximum eligibility. Cash bonuses paid do not exceed the maximum eligibility amount provided for under the table “Fiscal 2016 Grants of Plan-Based Awards”.
90
Our 2010 Performance Incentive Plan provides alternative forms of long-term incentives for our executive officers, including stock options with time and performance-based vesting, which require the market value of our common stock to increase before they are valuable. We do not use a targeted split of cash and equity when setting compensation for our executive officers.
The number of stock options granted is discretionary, but is based on our benchmarking principles described above. The value earned on any grant varies with the stock price over the option term. In large part due to the length of product development cycles, it is critical for our business to align the interests of executive officers and stockholders, and to retain executive officers by means of what we hope will be long-term wealth creation in the value of their stock options, which have vesting provisions that encourage continued employment. We elect to use stock options as a portion of our long-term equity incentive vehicle. Stock option grants are made at the commencement of employment, may be made annually and, occasionally, may be made following a significant change in job responsibilities or to meet other special objectives, including strategic goals and retention. Additionally, annual stock option grants typically occur the later of the filing of our annual report on Form 10-K for the most recently completed year, or the first trading day in which we are not in a blackout period, and are targeted around the 50th percentile of our peer group (in terms of market value), subject to adjustment for the compensation factors described above and the availability of equity under our equity-based compensation plans. We expect to continue to use stock options as a long-term incentive vehicle because:
|
|
• |
stock options align the interests of executives with those of the stockholders, support a pay-for-performance culture, foster employee stock ownership and focus the management team on increasing value for our stockholders; |
|
|
• |
stock options help to provide a balance to the overall executive compensation program as base salary and our cash incentive compensation program focus on nearer-term achievements, while the grant and vesting of stock options is intended to focus executive efforts towards increasing stockholder value over the longer term; |
|
|
• |
the vesting period of stock options encourages executive retention as long as the options remain in the money; and |
|
|
• |
we believe the use of stock options assists us in making our compensation package attractive to current and potential executive candidates, while conserving cash. |
Although stock option grants encourage equity ownership, we currently do not require our directors or executive officers to own a particular number of shares of our common stock. The Compensation Committee believes that stock, option and RSU holdings among our directors and executive officers are sufficient at this time to align this group’s interests with those of our stockholders.
In 2016, our executive officers located in Canada participated in the same group insurance and employee benefit plans as our other salaried employees in Canada and our executive officers located in the United States participate in the same group insurance and employee benefit plans as our other salaried employees in the United States. Tax preparation services are paid for executive officers who owe additional tax liabilities incurred by working in non-resident countries. At this time, we do not provide other special benefits or other perquisites to our executive officers.
Salary
Scott Cormack is our Chief Executive Officer and President. With respect to determining Mr. Cormack’s base salary for the 2016 fiscal year, the Compensation Committee considered his leadership in helping to develop the company’s product candidates and set the company’s strategic goals. The Compensation Committee also considered the fact that the AFFINITY trial did not meet its survival endpoint in a prospectively defined subpopulation of patients with poor prognosis, and that the final survival results for both the Phase 3 AFFINITY and the ENSPIRIT trials were expected to occur in 2016. It also conducted a comprehensive review, analysis and discussion of the salaries of executives as reported by Radford Executive Compensation Report in 2015. Based on these considerations, our Compensation Committee to leave Mr. Cormack’s base salary for 2016 unchanged at US$541,383, which is at the 75th percentile of our peer company survey from 2015.
Dr. Cindy Jacobs is our Executive Vice President and Chief Medical Officer. In determining her base salary for 2016, the Compensation Committee considered Dr. Jacobs’ important role in furthering the development of our clinical assets and extensive experience in obtaining U.S. Food and Drug Administration approval for oncology product candidates, as well as her instrumental role in partnering, alliance management and strategic discussions. The Compensation Committee also considered the fact that the
91
AFFINITY trial did not meet its survival endpoint in a prospectively defined subpopulation of patients with poor prognosis, and that the final survival results for both the Phase 3 AFFINITY and the ENSPIRIT trials were expected to occur in 2016. It also conducted a comprehensive review, analysis and discussion of the salaries of executives as reported by Radford in 2015. Based on these considerations, our Compensation Committee decided to leave Dr. Jacobs’ base salary for 2016 unchanged at $413,225, which is at the 75th percentile of our peer company survey from 2015.
John Bencich is our Vice President and Chief Financial Officer. In determining Mr. Bencich’s base salary for 2016, the Compensation Committee took into account his financial and accounting background and his corporate development expertise. The Compensation Committee also considered the fact that the AFFINITY trial did not meet its survival endpoint in a prospectively defined subpopulation of patients with poor prognosis, and further that the final survival results for both the Phase 3 AFFINITY and the ENSPIRIT trials were expected to occur in 2016. It also conducted a comprehensive review, analysis and discussion of the salaries of executives as reported by Radford in 2015. Based on these considerations, our Compensation Committee decided to leave Mr. Bencich’s base salary unchanged for 2016 at $307,500, which is at the 50th percentile of our peer company survey from 2015.
Cash Incentives
In accordance with the Compensation Committee’s goal to target compensation around the 50th percentile of our peer group as described further in the section entitled “Benchmarking of Executive Compensation” and based on a comprehensive review, analysis and discussion of the Radford report, each Named Executive Officer’s bonus potential for 2016, was as follows:
|
Executive Officer |
Short-Term Incentive Award Eligibility |
|
Scott Cormack, Chief Executive Officer and President |
55% of salary |
|
Cindy Jacobs, Chief Medical Officer and Executive Vice President |
40% of salary |
|
John Bencich, Vice President and Chief Financial Officer |
40% of salary |
In the first quarter of 2016, the Board of Directors adopted the following 2016 corporate objectives under the STIP:
|
Corporate Objectives |
Weighting |
|
Optimize Custirsen’s potential value by facilitating timely phase 3 data analysis and release
|
35% |
|
Optimize Apatorsen’s potential value by facilitating timely phase 2 data analysis and release and defining a bladder cancer development strategy
|
30% |
|
Continuously assess strategic alternatives to support or accelerate the corporate mission
|
35% |
The Board of Directors selected these particular corporate objectives based on its judgment that they represented areas over which the Named Executive Officers have significant operational control and on which the Board of Directors believed they should focus to move our strategic plan forward and enhance stockholder value during 2016. The total weighting of corporate objectives under the 2016 STIP was a target of 100%.
Performance Against 2016 Corporate Objectives
Notwithstanding that the executive officers facilitated timely phase 3 data analysis and release related to the custirsen clinical trials, as well as phase 2 data analysis and release related to the apatorsen clinical trials, both the AFFINTIY and ENSPIRIT clinical trials did not meet their primary endpoint of extending survival with statistical significance. Further, while a merger agreement was announced in January 2017, it was not completed in 2016. Accordingly, the Compensation Committee exercised its discretion and determined not to award any bonuses for performance of the 2016 corporate objectives.
92
Stock option grants are discretionary based on the Compensation Committee’s analysis of employee achievement of company-wide and individual objectives and our benchmarking principles. The Compensation Committee determined to award each Named Executive Officer non-qualified stock options to acquire shares of our common stock pursuant to the terms and conditions of the 2010 Performance Incentive Plan. In each case, the Compensation Committee considered and evaluated the Radford report, the compensation factors described above, our stock price and the amount of equity available for grant under our 2010 Performance Incentive Plan.
The number of stock options granted was calculated using an option grant dollar value (based on a Black-Scholes model) and was compared against the percentage of total equity ownership in order to ensure that the recommendations were within the benchmarks described below. The options were performance-based with 50% of the shares vesting upon the achievement by December 31, 2016 of the earlier to occur of (i) positive results from either the AFFINITY or ENSPIRIT trials, or (ii) consummation of a change in control. If one of the milestones were met, the remaining 50% would vest monthly over two years. Each option was granted with a ten-year term and an exercise price equal to the closing price of our common stock on NASDAQ on the date of grant. As no milestones were achieved by December 31, 2016, the options were cancelled.
The Compensation Committee determined not to grant RSUs or establish performance RSUs for 2016 on the basis that it would not provide appropriate incentive for the material milestones expected to occur in 2016.
Advisory Vote on Executive Compensation
In 2011 and 2014, we held advisory say-on-pay votes to approve the compensation of our executive officers, with approximately 98% and 95%, respectively, of the votes cast in favor of our executive compensation program. In light of the support by our stockholders of our executive compensation program, the Compensation Committee did not make any significant changes to our 2015 executive compensation program.
In addition, at our 2011 annual meeting of stockholders, a majority of our stockholders voted in favor of an advisory vote on executive compensation every three years, and, accordingly, our Board of Directors determined that we will hold an advisory vote on executive compensation at our annual meeting of stockholders every three years. We expect to hold the next vote on executive compensation at the 2017 annual meeting of stockholders.
Other Policies and Considerations
Internal Pay Equity
The Compensation Committee reviewed the 2015 Radford report on compensation and concluded that total compensation for a company’s chief executive officer is generally higher than the total compensation for either its chief financial officer or chief medical officer, and that the total compensation of the chief medical officer is generally higher than the total compensation of the chief financial officer. The relative total compensation for our executive officers for 2016 followed the same pattern observed in the Radford report, with our Chief Executive Officer receiving the highest total compensation, followed by our Chief Medical Officer and then our Vice President and Chief Financial Officer. Our ordinal pay ranking is consistent with comparable companies, and as each component of compensation for each executive officer is determined in relation to the 50th to 75th percentile of officers holding positions having similar responsibilities at comparable companies, the Compensation Committee believes that relative compensation among our executive officers is appropriate and consistent with maintaining internal pay equity.
Relationship Between Compensation Elements
Each element of executive officer compensation was determined with reference to the 50th to 75th percentile of the same element paid to executive officers holding the similar position at comparable companies. Therefore, no objective formula was utilized when determining the relative proportion of salary, cash incentive or equity awards relative to each other or to total compensation.
Employment Agreements and Termination Benefits
The employment agreement for each executive officer contains provisions related to termination and change of control. When establishing the termination and change of control provisions of the employment agreements, the Compensation Committee and the Board of Directors considered the Radford report, which provided recommendations to the Compensation Committee regarding the termination and change of control provisions for each executive officer based on publicly available information regarding the practices of our peer group, policy statements made by significant investor groups and an analysis of current market trends. We provide change in control protections to our officers to alleviate concerns regarding the possible occurrence of such a transaction, allowing them to
93
focus their attention on our business. In addition, these protections encourage executives to remain with us during the threat or negotiation of a change in control transaction, which preserves our value and the potential benefit to be received by our stockholders in the transaction. The specific terms of the termination and change of control arrangements, as well as an estimate of the compensation that would have been payable had they been triggered as of the end of 2016, are described in detail in the section below entitled “Executive Compensation—Potential Cost of Termination Payments.”
Compensation Committee Interlocks and Insider Participation
No member of the Compensation Committee during fiscal year 2016 served as one of our officers, former officers or employees nor received directly or indirectly compensation from the Company, other than in the capacity as a member of our Board and Compensation Committee. There was no direct or indirect control by the members of the Compensation Committee of the Company. No member of the Compensation Committee, directly or indirectly, is the beneficial owner of more than 10% of the Company’s equity, nor are they an executive officer, employee, director, general partner or a managing member of one or more entities that are together the beneficial owners of more than 10% of the Company’s equity. The Compensation Committee members are not aware of any business or personal relationship between (i) a member of the Compensation Committee and any person who has provided or is providing advice to the Compensation Committee; and (ii) an executive officer of the company and any firm or other person who is employed or is employing such person to provide advice to the Compensation Committee. During fiscal year 2016, none of our executive officers served as a member of the compensation committee of any other entity, one of whose executive officers served as a member of our Board of Directors or Compensation Committee, and none of our executive officers served as a member of the board of directors of any other entity, one of whose executive officers served as a member of our Compensation Committee.
The Compensation Committee of the Board of Directors has reviewed and discussed “Compensation Discussion and Analysis” required by Item 402(b) of Regulation S-K with management and, based on such review and discussions, the Compensation Committee recommended to the Board of Directors that the “Compensation Discussion and Analysis” be included in the our 2016 Annual Report on Form 10-K and this Proxy Statement.
COMPENSATION COMMITTEE OF THE BOARD OF DIRECTORS
Neil Clendeninn, Chairperson
Jack Goldstein
Martin Mattingly
The information contained in the report above shall not be deemed to be “soliciting material” or to be “filed” with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933, as amended, except to the extent specifically incorporated by reference in such filing.
During the 2016 fiscal year, our Named Executive Officers and their respective positions were as follows: Scott Cormack, Chief Executive Officer, President, Treasurer and Secretary; Cindy Jacobs, Ph.D., M.D., Executive Vice President and Chief Medical Officer; and John Bencich, Vice President and Chief Financial Officer.
Mr. Cormack, Dr. Jacobs and Mr. Bencich are referred to as our Named Executive Officers for purposes of this Proxy Statement.
94
The following table sets forth information regarding the compensation of our Named Executive Officers for each of the fiscal years ended December 31, 2016, 2015 and 2014. The components of the compensation reported in the Summary Compensation Table are described below. Additional information on the components of the total compensation package, including a discussion of the proportion of each element to total compensation, is discussed in “Compensation Discussion and Analysis.”
|
Name and Principal Position
|
Year
|
|
Salary ($)
|
|
Bonus ($)
|
|
Stock Awards ($)(1)
|
|
Option Awards ($)(1)
|
|
Non-Equity Incentive Plan Compensation ($)
|
|
Total ($)
|
|
Scott Cormack, President and Chief Executive Officer
|
2016 2015 2014
|
|
541,383(2) 541,383(3) 497,153(4)
|
|
— — —
|
|
— 69,750 655,320
|
|
155,202 79,473 410,321
|
|
— 148,890 156,324
|
|
696,585 839,496 1,719,118
|
|
Cindy Jacobs, Executive Vice President and Chief Medical Officer
|
2016 2015 2014
|
|
413,225 413,225 413,225
|
|
— — —
|
|
— 34,875 348,325
|
|
69,840 39,737 205,161
|
|
— 82,650 86,777
|
|
483,065 570,487 1,053,488
|
|
John Bencich,
|
2016 2015 2014
|
|
307,500 307,500 118,269(5)
|
|
— — 20,000
|
|
— 23,250 63,400
|
|
58,200 26,491 93,328
|
|
— 53,820 18,512
|
|
365,700 411,061 313,509
|
|
(1) |
The dollar amounts in this column reflect the aggregate grant date fair value of equity awards granted during the fiscal year in accordance with FASB Accounting Standards Codification Topic 718 for stock-based compensation. For performance-based RSUs awarded in 2014, the dollar amounts also reflect the value at the grant date based upon the probable outcome of such conditions. These amounts do not correspond to the actual cash value that will be recognized by each of the Named Executive Officers when received. For a discussion of the assumptions and methodologies used to value the awards reported in this column, see note 10 to our audited consolidated financial statements, which are included in its 2016 Annual Report on Form 10-K. During 2014, 32,000 and 20,000, performance RSUs were forfeited by Mr. Cormack and Dr. Jacobs, respectively, as the SYNERGY trial did not meet its primary endpoint. In 2016, the 2016 options awards included in this column were cancelled for each of Mr. Cormack, Dr. Jacobs and Mr. Bencich. These options vested upon either the positive results from the AFFINITY or ENSPIRIT trials, or the consummation of a change in control by December 31, 2016. Neither of these milestones were achieved, and therefore, all of the options granted to Mr. Cormack, Dr. Jacobs and Mr. Bencich in 2016 were cancelled. Additionally, in 2016, 8,000 and 5,000 performance RSUs were forfeited by Mr. Cormack and Dr. Jacobs, respectively, as the AFFINITY trial did not meet its primary endpoint. |
|
(2) |
From January 1, 2016 to June 15, 2016, Mr. Cormack’s salary was paid as to 25% in Canadian dollars of CDN$83,733 and 75% in U.S. dollars of $186,100. From June 16, 2016 to December 31, 2016, Mr. Cormack’s salary was paid as to 50% in Canadian dollars of CDN$192,046 and 50% in U.S. dollars of $146,624. The portion paid in Canadian dollars was converted using the monthly average noon foreign exchange rate from the prior month for the current payment date, which resulted in an average exchange rate of US$1.00 = CDN$1.3216. |
|
(3) |
For 2015, Mr. Cormack’s salary was paid as to 25% in Canadian dollars of CDN$171,720 and 75% in U.S. dollars of $406,037. The portion paid in Canadian dollars was converted using the noon spot foreign exchange rate at each payment date, which resulted in an average exchange rate of US$1.00 = CDN$1.2764. |
|
(4) |
For fiscal year 2014, Mr. Cormack’s salary was paid as to 25% in Canadian dollars of CDN$138,052 and 75% in U.S. dollars of $372,161. The portion paid in Canadian dollars was converted using the 2014 average annual foreign exchange rate of US$1.00 = CDN$1.1045. |
|
(5) |
Reflects Mr. Bencich’s salary from the commencement of his employment on August 11, 2014 through December 31, 2014 |
95
Fiscal 2016 Grants of Plan-Based Awards
The following table provides information related to grants of plan-based awards to our Named Executive Officers during the 2016 fiscal year.
|
|
|
Estimated |
|
|
|
|
|
|
Name |
Grant |
Target(1) ($) |
|
|
All Other Option Awards: # of Securities Underlying Options (2) |
Exercise or Base Price of Option Awards ($/Sh) |
Grant Date Fair Value of Stock and Option Awards (3)($) |
|
Scott Cormack |
3/14/2016 |
|
|
|
300,000 |
0.85 |
155,202 |
|
|
|
297,760 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cindy Jacobs |
3/14/2016 |
|
|
|
135,000 |
0.85 |
69,840 |
|
|
|
165,290 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
John Bencich |
3/14/2016 |
|
|
|
112,500 |
0.85 |
58,200 |
|
|
|
123,000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(2) |
The amounts shown in this column represent stock options granted under our 2010 Performance Incentive Plan, which have subsequently been cancelled as none of the performance milestones were achieved by December 31, 2016. |
|
(3) |
Amounts represent the grant date fair value of stock option awards measured in accordance with the guidance in FASB ASC Topic 718. These amounts do not correspond to the actual cash value that will be recognized by each of the Named Executive Officers when received. For a discussion of the assumptions and methodologies used to value the awards reported in this column, see note 10 to our audited consolidated financial statements, which are included in this 2016 Annual Report on Form 10-K. |
96
Outstanding Equity Awards at Fiscal Year-End
The following table provides information regarding the outstanding equity awards held by the Named Executive Officers as of December 31, 2016.
|
|
|
OPTION AWARDS |
|
Stock Awards |
||||||||||||||
|
|
|
|
|
|
|
|||||||||||||
|
Name |
|
Number of |
|
Number of |
|
|
Option |
|
Option |
|
Equity Incentive Plan Awards: Number of Unearned Shares, Units or Other Rights that Have Not Vested (#) |
|
Equity Incentive Plan Awards: Market or Payout Value of Unearned Shares, Units or Other Rights that Have Not Vested ($) |
|
Number of Shares or Units or Other Rights that Have |
|
Market Value of Shares or Units or Other Rights that Have ($) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Scott Cormack, President and Chief Executive Officer |
|
25,000 40,000 37,500 36,719 36,454 35,937 |
|
— — — 781 13,546 39,063 |
|
|
22.28 15.97 13.00 11.95 1.86 |
|
12/31/19 12/14/20 05/08/22 03/12/23 03/14/24 05/19/25 |
(1) (3) (4) (5) (6) |
|
|
|
|
4,687 12,500 18,750 28,125
|
(7) (8) (9) (10) |
56,010 147,375 66,563 52,313 |
|
|
Cindy Jacobs, Executive Vice President and Chief Medical Officer |
|
12,000 17,969
|
|
— — — 417 6,773 19,531 |
|
|
22.28 15.97 11.95 11.79 1.86 |
|
12/31/19 12/14/20 03/12/23 03/14/24 05/19/25 |
(1) (5) (6) |
|
|
|
|
2,500 6,250 10,000 14,062
|
(7) (8) (9) (10) |
29,875 73,688 35,500 26,155 |
|
|
John Bencich, Vice President and Chief Financial Officer |
|
23,333 11,979 |
|
16,667 13,021
|
|
|
3.17 1.86
|
|
08/12/24 05/19/25
|
(11) (6)
|
|
|
|
|
10,000 9,375
|
(12) (10) |
31,700 17,438 |
|
________________________________________
|
(1) |
These stock options were granted under the 2007 Performance Incentive Plan and were fully vested on December 31, 2013. |
|
(2) |
These stock options were granted under the 2010 Performance Incentive Plan and were fully vested on December 31, 2014. |
|
(3) |
These stock options were granted under the 2010 Performance Incentive Plan and were fully vested on December 31, 2016. |
|
(4) |
These stock options were granted under the 2010 Performance Incentive Plan and vest monthly over a 48-month period beginning January 1, 2013. |
|
(5) |
These stock options were granted under the 2010 Performance Incentive Plan and vest monthly over a 48-month period beginning January 1, 2014. |
|
(6) |
These stock options were granted under the 2010 Performance Incentive Plan and vest monthly over a 48-month period beginning January 1, 2015. |
|
(7) |
These RSUs were granted under the 2010 Performance Incentive Plan and vest annually over four years beginning January 1, 2013. |
|
(8) |
These RSUs were granted under the 2010 Performance Incentive Plan and vest annually over four years beginning January 1, 2014. |
|
(9) |
These RSUs were granted under the 2010 Performance Incentive Plan and vest annually over four years beginning June 12, 2014. |
|
(10) |
These RSUs were granted under the 2010 Performance Incentive Plan and vest annually over four years beginning January 1, 2015. |
|
(11) |
These stock options were granted under the 2010 Performance Incentive Plan and vest monthly over a 48-month period beginning August 12, 2014. |
|
(12) |
These RSUs were granted under the 2010 Performance Incentive Plan and vest annually over four years beginning August 12, 2014. |
97
Option Exercises and Stock Vested
The following table provides information related to the vesting of RSUs held by the Named Executive Officers during the 2016 fiscal year.
|
|
Stock Awards |
|||||||
|
|
Number of Shares Acquired on Vesting |
|
Value Realized |
|||||
|
Name |
(#) |
|
($) |
|||||
|
Scott Cormack, President and Chief Executive Officer
Cindy Jacobs, Executive Vice President and Chief Medical Officer
John Bencich, Vice President Chief Financial Officer
|
12,967 4,519
8,934 3,563
2,291 3,620
|
(1) (2)
(1) (2)
(1) (3)
|
10,503.28 4,880.52
7,236.53 3,848.04
1,855.71 1,991.00
|
|||||
|
(1) |
These RSUs were granted under the 2010 Performance Incentive Plan and vested on March 14, 2016. |
|
(2) |
These RSUs were granted under the 2010 Performance Incentive Plan and vested on June 12, 2015. |
|
(3) |
These RSUs were granted under the 2010 Performance Incentive Plan and vested on August 18, 2015. |
Pension Benefits/Nonqualified Deferred Compensation
We do not have any plan that provides for payments or other benefits at, following, or in connection with retirement. We also do not have a plan that provides for the deferral of compensation for any employee.
Potential Payments Upon Termination/Change of Control
Change of Control Under Our Equity Compensation Plans
The following discussion sets forth the change of control provisions provided for in our various equity compensation plans.
2007 Performance Incentive Plan
Under the 2007 Performance Incentive Plan, or the 2007 Plan, the administrator has the discretion to provide in each award agreement the terms and conditions with respect to a change of control that relate to (1) the vesting of an award and (2) the assumption of an award or issuance of comparable securities under an incentive program. If the terms of an option agreement provide for accelerated vesting in the event of a change of control, or to the extent that an option is vested and not yet exercised, the administrator may provide for the purchase or exchange of each option for an amount of cash or other property. Outstanding options shall terminate and cease to be exercisable upon a change of control except to the extent that the options are assumed by the successor entity, or parent of the successor entity, pursuant to the terms of the change of control transaction.
As used in the 2007 Plan, the term “change of control” means the occurrence of any of the following:
|
|
• |
acquisitions of our securities possessing more than 50% of the total combined voting power of all of our outstanding securities; |
|
|
• |
a merger or consolidation with any other entity, whether or not we are the surviving entity in such transaction, except for a transaction in which the holders of our outstanding voting securities immediately prior to such merger or consolidation hold, as a result of holding our securities prior to such transaction, in the aggregate, securities possessing more than 50% of the total combined voting power of all of our outstanding voting securities or the voting securities of the surviving entity, or the parent of the surviving entity, immediately after such merger or consolidation; |
|
|
• |
the sale, transfer or other disposition, in one transaction or a series of related transactions, of all or substantially all of our assets; or |
|
|
• |
the approval by our stockholders of a plan or proposal for our liquidation or dissolution. |
98
2010 Performance Incentive Plan
Under the 2010 Plan, the administrator has the discretion to provide in each award agreement the terms and conditions with respect to a change of control that relate to the vesting of an award and the assumption of an award or issuance of comparable securities under an incentive program. If the terms of an option agreement provide for accelerated vesting in the event of a change of control, or to the extent that an option is vested and not yet exercised, the administrator may provide for the purchase or exchange of each option for an amount of cash or other property. Outstanding options shall terminate and cease to be exercisable upon a change of control except to the extent that the options are assumed by the successor entity (or parent of the successor entity) pursuant to the terms of the change of control transaction. As used in the 2010 Plan, the term “change of control” means the occurrence of any of the following:
|
|
• |
acquisitions of our securities possessing more than 50% of the total combined voting power of all of our outstanding securities; |
|
|
• |
a merger or consolidation with any other entity, whether or not we are the surviving entity in such transaction, except for a transaction in which the holders of our outstanding voting securities immediately prior to such merger or consolidation hold, as a result of holding our securities prior to such transaction, in the aggregate, securities possessing more than 50% of the total combined voting power of all of our outstanding voting securities or the voting securities of the surviving entity, or the parent of the surviving entity, immediately after such merger or consolidation; |
|
|
• |
the sale, transfer or other disposition, in one transaction or a series of related transactions, of all or substantially all of our assets; or |
|
|
• |
the approval by the stockholders of a plan or proposal for our liquidation or dissolution. |
Termination and Change of Control Provisions Under Employment Agreements
As of December 31, 2016, we have employment agreements in place with each of our Named Executive Officers that provide for compensation upon the termination of their employment under certain circumstances, as described below.
Cormack Agreement
The agreement between us, OncoGenex Technologies Inc. and Mr. Cormack, which is referred to as the Cormack Agreement, provides Mr. Cormack with termination benefits in the event Mr. Cormack is terminated without cause or for disability, or if Mr. Cormack resigns for good reason, or Good Reason, which means due to (i) the relocation of the officer’s primary work location by more than 40 miles from the current office location; (ii) a material reduction of the officer’s base salary or employee benefits; (iii) any material reduction or diminution of the officer’s duties, authority or responsibilities; (iv) a fundamental breach by us of the Cormack Agreement; or (v) the failure of any successor to assume expressly in writing our obligations under the Cormack Agreement, in each case, provided that Mr. Cormack has provided us with two months’ advance written notice and an opportunity to cure such breach during such two-month period. Any termination that occurs without cause, due to disability or for Good Reason is referred to as an Involuntary Termination.
The Cormack Agreement provides that if an Involuntary Termination occurs, we will be obligated to pay Mr. Cormack a lump sum equal to 18 months of his then-current base salary. In addition, Mr. Cormack will receive continued entitlement under group medical, dental and insurance plans, excluding short- and long-term disability plans and pension plans, to which Mr. Cormack and his family are entitled at Mr. Cormack’s termination date, to the extent such benefit plans permit for 18 months or until Mr. Cormack becomes employed elsewhere where comparable benefits are provided, whichever date comes first (this period is referred to as the Cormack Benefit Plan Severance Period). To the extent continuance of a benefit plan, excluding short- and long-term disability plans and pension plans, is not permitted, OncoGenex Technologies Inc. will be obligated to pay Mr. Cormack an amount equal to the sum Mr. Cormack would be required to pay to receive comparable benefits for the Cormack Benefit Plan Severance Period. Notwithstanding the terms of any of our equity compensation plans or any agreement in connection with such plans, if there is an Involuntary Termination, then the time-based vesting restrictions, if any, will immediately lapse on an additional number of shares under all of Mr. Cormack’s outstanding compensatory equity awards, which includes any outstanding stock options granted to Mr. Cormack under our equity compensation plans, that would have time-vested if Mr. Cormack had continued his employment for 18 months following his Involuntary Termination.
The Cormack Agreement provides for additional termination benefits if an Involuntary Termination occurs during the period beginning three months before and ending 12 months after a change in control or if such Involuntary Termination is required by the merger agreement, purchase agreement or other instrument relating to such change in control or such Involuntary Termination is made at the express request of the other party or parties to the transaction constituting such change in control, each of which events is referred to as a Change in Control Termination. Upon a Change in Control Termination, we will be obligated to pay Mr. Cormack 24 months of his then-current base salary, plus a sum equal to 12 months of his average monthly bonus earnings, where such average is
99
calculated over the 24-month period immediately preceding Mr. Cormack’s termination date and based on Mr. Cormack’s bonuses paid in such 24-month period. In addition, Mr. Cormack will receive continued entitlement under our benefit plans as described above, or an amount equal to the sum Mr. Cormack would be required to pay to receive comparable benefits if such continued entitlement is not permitted as described above, except that the Cormack Benefit Plan Severance Period will be 24 months instead of 18 months. Notwithstanding the terms of any of our equity compensation plans or any agreement in connection with such plans, upon a Change in Control Termination, all vesting restrictions, if any, will immediately lapse on all of Mr. Cormack’s compensatory equity awards effective as of his termination date.
All termination benefits in the event of an Involuntary Termination or Change in Control Termination are subject to Mr. Cormack’s execution, delivery and non-revocation of a general release of all litigation and other claims against us and our affiliates.
Mr. Cormack’s employment will terminate upon the consummation of our planned merger. Upon such termination, Mr. Cormack will receive the benefits he’s entitled to upon an Involuntary Termination upon a Change in Control Termination.
Jacobs Agreement
Our agreement with Cindy Jacobs, referred to as the Jacobs Agreement, provides Dr. Jacobs with termination benefits in the event of an Involuntary Termination, provided that, in the case of termination for good reason, Dr. Jacobs has provided us with 30 days’ advance written notice and an opportunity to cure such breach during such 30-day period.
The Jacobs Agreement provides that if an Involuntary Termination occurs, we will be obligated to pay Dr. Jacobs a lump sum payment equal to 12 months of her then-current base salary. In addition, if Dr. Jacobs elects to continue her and her dependents’ health insurance coverage under COBRA, we must pay up to 12 months of Dr. Jacobs’ monthly premium under COBRA, provided that our obligation to pay the monthly premium will cease when Dr. Jacobs becomes eligible to receive substantially equivalent health coverage in connection with new employment. Notwithstanding the terms of any of our equity compensation plans or any agreement in connection with such plans, if there is an Involuntary Termination, then the time-based vesting restrictions, if any, will immediately lapse on an additional number of shares under all of Dr. Jacobs’ outstanding compensatory equity awards, which includes outstanding stock options granted to Dr. Jacobs under our equity compensation plans, that would have time-vested if Dr. Jacobs had continued in employment for 12 months following her Involuntary Termination.
The Jacobs Agreement provides for additional termination benefits if an Involuntary Termination occurs during the period beginning three months before and ending 12 months after a change in control or if such Involuntary Termination is required by the merger agreement, purchase agreement or other instrument relating to such change in control, or such Involuntary Termination is made at the express request of the other party or parties to the transaction constituting such change in control, each of which events is referred to as a Change in Control Termination. Upon such a Change in Control Termination, we will be obligated to pay Dr. Jacobs 15 months of her then-current base salary, plus a sum equal to 12 months of her average monthly bonus earnings, where such average is calculated over the 24-month period immediately preceding Dr. Jacobs’ separation from services and based on Dr. Jacobs’ bonuses paid in such 24-month period. In addition, our payment of monthly COBRA premiums as described above will be for up to 15 months instead of up to 12 months. Notwithstanding the terms of any of our equity compensation plans or any agreement in connection with such plans, upon a Change in Control Termination, all vesting restrictions, if any, will immediately lapse on all of Dr. Jacobs’ compensatory equity effective as of her separation from service.
All termination benefits in the event of an Involuntary Termination or Change in Control Termination are subject to Dr. Jacobs’ execution, delivery and non-revocation of a general release of all litigation and other claims against us and our affiliates.
Bencich Agreement
Our agreement with John Bencich, referred to as the Bencich Agreement, provides Mr. Bencich with termination benefits in the event of an Involuntary Termination, provided that, in the case of termination for good reason, Mr. Bencich has provided us with 30 days’ advance written notice and an opportunity to cure such breach during such 30-day period. We may terminate the Agreement with or without cause by giving Mr. Bencich 30 days’ advance written notice, or a cash payment equivalent to 30 calendar days of his then-current base salary in lieu of providing such notice.
The Bencich Agreement provides that if an Involuntary Termination occurs, we will be obligated to pay Mr. Bencich a lump sum payment equal to 12 months of his then-current base salary. In addition, if Mr. Bencich elects to continue his and his dependents’ health insurance coverage under COBRA, we must pay in a lump sum payment the number of months of Mr. Bencich’s monthly premium under COBRA, that is equal to the 12 months. Notwithstanding the terms of any of our equity compensation plans or any agreement in connection with such plans, if there is an Involuntary Termination, then the time-based vesting restrictions, if any, will immediately lapse on an additional number of shares under all of Mr. Bencich’s outstanding compensatory equity awards, which
100
includes outstanding stock options granted to Mr. Bencich under our equity compensation plans, that would have time-vested if Mr. Bencich had continued in employment for 12 months following his Involuntary Termination.
The Bencich Agreement provides for additional termination benefits if an Involuntary Termination occurs during the period beginning three months before and ending 12 months after a change in control or if such Involuntary Termination is required by the merger agreement, purchase agreement or other instrument relating to such change in control, or such Involuntary Termination is made at the express request of the other party or parties to the transaction constituting such change in control, each of which events is referred to as a Change in Control Termination. Upon such a Change in Control Termination, we will be obligated to pay Mr. Bencich 15 months of his then-current base salary, plus a sum equal to 15 months of his average monthly bonus earnings, where such average is calculated over the 24-month period immediately preceding Mr. Bencich’s separation from services and based on Mr. Bencich’s bonuses paid in such 24-month period. In addition, our payment of monthly COBRA premiums as described above will be for up to 15 months instead of up to 12 months. Notwithstanding the terms of any of our equity compensation plans or any agreement in connection with such plans, upon a Change in Control Termination, all vesting restrictions, if any, will immediately lapse on all of Mr. Bencich’s compensatory equity effective as of his separation from service.
All termination benefits in the event of an Involuntary Termination or Change in Control Termination are subject to Mr. Bencich’s execution, delivery and non-revocation of a general release of all litigation and other claims against us and our affiliates.
Potential Cost of Termination Payments
In the table below, we have estimated the potential cost to us of the compensation to which each Named Executive Officer would have been entitled if he or she experienced an Involuntary Termination or a Change in Control Termination effective as of December 31, 2016.
|
Named |
Involuntary Termination |
Involuntary Termination in Connection |
||||||
|
Cash Payments ($) |
Benefits ($) |
Equity Compensation ($) (2) |
Total ($) |
Cash |
Benefits ($) |
Equity Compensation ($) |
Total ($) |
|
|
Scott Cormack(1) |
812,085 |
66,047 |
451,390(3) |
1,329,522 |
1,157,225 |
88,063 |
474,816(4) |
1,720,104 |
|
Cindy Jacobs |
413,225 |
16,342 |
206,236(5)
|
635,803 |
557,862 |
20,428 |
241,495(6) |
819,785 |
|
John Bencich |
307,500 |
38,533 |
57,970(7) |
404,003 |
411,285 |
48,167 |
101,822(8) |
561,274 |
|
(1) |
Mr. Cormack’s employment will terminate upon the consummation of our planned merger. Upon such termination, Mr. Cormack will receive the benefits he’s entitled to upon an Involuntary Termination upon a Change in Control Termination. |
|
(2) |
The employment agreements for each of Mr. Cormack, Dr. Jacobs, and Mr. Bencich state that the time-based vesting restrictions associated with unvested options immediately lapse on any shares of common stock that would have time-vested if they had had continued in employment throughout their respective severance period as defined in their employment agreements. The amounts above represent the stock option expense that would be incurred by us in accordance with the guidance of FASB ASC Topic 718 in relation to options vested immediately upon termination in accordance with the terms of the individual’s employment agreement. |
|
(3) |
Represents stock option expense associated with the accelerated vesting of 781 options with an exercise price of $11.95 per share and 13,546 options with an exercise price of $11.79 per share and 28,126 options with an exercise price of $1.86 per share and 57,031 accelerated RSUs. |
|
(4) |
Represents stock option expense associated with the accelerated vesting of 781 options with an exercise price of $11.95 per share, 13,546 options with an exercise price of $11.79 per share and 37,501 options with an exercise price of $1.86 per share and 64,062 accelerated RSUs. |
|
(5) |
Represents stock option expense associated with the accelerated vesting of 417 options with an exercise price of $11.95 per share, 6,252 options with an exercise price of $11.79, and 9,375 options with an exercise price of $1.86 per share and 22,448 accelerated RSUs. |
|
(6) |
Represents stock option expense associated with the accelerated vesting of 417 options with an exercise price of $11.95 per share, 6,773 options with an exercise price of $11.79, and 11,719 options with an exercise price of $1.86 per share and 32,812 accelerated RSUs. |
|
(7) |
Represents stock option expense associated with the accelerated vesting of 10,000 options at an exercise price of $3.17 per share and 6,250 options with an exercise price of $1.86 per share and 10,688 accelerated RSUs. |
|
(8) |
Represents stock option expense associated with the accelerated vesting of 12,500 options at an exercise price of $3.17 per share and 7,813 options with an exercise price of $1.86 per share and 19,375 accelerated RSUs. |
Director Compensation
Overview
The charter of the Compensation Committee provides that the Compensation Committee is to recommend to the Board of Directors matters related to director compensation. The director compensation package for non-employee directors consists of annual cash
101
compensation and an award of restricted stock units and stock options exercisable to purchase shares of our common stock. None of our employees are entitled to receive compensation for service as a director. Our director compensation policy for fiscal year 2016 is set forth below under the heading “Director Compensation Policy – 2016 Director Compensation”.
Peer Group Used for Benchmarking 2016 Compensation
Our management regularly reviews peer group data compiled by Radford to determine whether total direct compensation and each component of the compensation package are approximately equal to the targeted 50th percentile for director compensation of our peer company list. Based on this review, management makes recommendations to the Compensation Committee that they deem necessary to align director compensation with the foregoing peer group target. The peer group companies are amended from time to time at the discretion of the Board of Directors.
The evaluation of peer group companies for purposes of establishing director compensation for fiscal year 2016 occurred in June 2015. In consideration of our market capitalization and number of employees at that time, the Compensation Committee recommended, and the Board of Directors approved, a peer group based on the following characteristics:
|
|
• |
late stage pre-commercial biotechnology/biopharmaceutical companies at a similar stage of drug development (Phase II to Phase III) |
|
|
• |
companies located in biotechnology hub markets (Seattle, San Francisco, San Diego and Boston) to reflect the recruiting market for executive talent; |
|
|
• |
companies with market values |
|
|
|
|
Research and development spending, available cash and enterprise value were also examined as additional metrics to help determine appropriate peer companies.
Based on the foregoing criteria, and the recommendations of Radford, the Compensation Committee recommended, and the Board of Directors approved, the following peer group for purposes of establishing our current director compensation policy:
|
Actinium Pharmaceuticals, Inc. |
MediciNova, Inc. |
|
Anthera Pharmaceuticals, Inc. |
MEI Pharma, Inc. |
|
ArQule Inc. |
Onconova Therapeutics, Inc. |
|
Aveo Pharmaceuticals, Inc. |
Oncothyreon Inc. |
|
BIND Therapeutics, Inc. |
OXiGENE, Inc. |
|
Celator Pharmaceuticals, Inc. |
Rexahn Pharmaceuticals, Inc. |
|
Celsion Corporation |
|
|
Conatus Pharmaceuticals Inc. |
Sunesis Pharmaceuticals, Inc. |
|
Cyclacel Pharmaceuticals, Inc. |
Targacept, Inc. |
|
|
TetraLogic Pharmaceuticals Corporation |
|
GTx, Inc. |
Threshold Pharmaceuticals, Inc. |
|
Kalobios Pharmaceuticals, Inc. |
Vical Incorporated |
2016 Director Compensation Policy
As part of its evaluation of compensation levels for the 2016 fiscal year, the Compensation Committee recommended and the Board of Directors approved the retention of Radford to review compensation levels of our independent directors and committee members. Radford was instructed to benchmark and make recommendations regarding the initial and annual retainer amounts for directors and chairpersons of the Board of Directors and the various committees, as well as the amounts and terms of initial and annual long-term equity incentive awards for directors. As a result in the decline of our market capitalization and based on peer group analyses, the Board of Directors decided that the compensation for non-employee directors in connection with their service on the Board of Directors and its committees would not be increased for 2016. Accordingly, 2016 director compensation policy was as follows:
|
|
• |
An annual retainer of $65,000 was paid to the Chairperson of the Board of Directors or the lead director and $40,000 was paid to all other non-employee directors, together with an excess meeting fee of $2,000 for each meeting held over 10 annual meetings. These retainers were paid in four quarterly installments. Each quarterly payment was conditioned on the director remaining a director on the date of actual payment, which was typically within 10 days following the completion of the respective calendar quarter. |
102
|
|
Chairperson |
Other Members |
|
Audit Committee |
$20,000 |
$10,000 |
|
Compensation Committee |
$15,000 |
$7,500 |
|
Nominating and Governance Committee |
$10,000 |
$5,000 |
|
|
• |
Any new director would have received a one-time initial grant of stock options to acquire 8,000 shares of our common stock upon becoming a director, which would vest over three years, with one-third vesting at each of the first, second and third anniversaries of the date of grant. In addition, any new director also would have received an initial grant of restricted stock units, or RSUs, to acquire 4,000 shares of our common stock which would vest over three years. |
|
|
• |
Each director that was re-elected by our stockholders at an annual meeting was to receive a grant of stock options to acquire 5,000 shares of common stock and RSUs to acquire 2,500 shares of common stock, with the Chairman of the Board receiving additional RSUs to acquire 2,500 shares of common stock, promptly following re-election. However, in May 2016, the Compensation Committee and Board determined not to grant RSUs for 2016 in consideration of the material milestones expected to occur in 2016 and our stock price. Accordantly, the Board granted all non-employee members of the Board options to purchase 22,500 shares of common stock, except for our Board Chairman, who received options to purchase 31,500 shares of common stock. Any director who was appointed or elected to the Board of Directors for the first time, and who received an initial stock option and RSU grant, would have received a first annual stock option and RSU grant in an amount equal to the product of the number of shares of common stock that would otherwise be subject to such annual stock option and RSU grant multiplied by the fraction of a year during which the director served on the Board of Directors immediately preceding the date of the annual meeting. Each annual stock option and RSU grant vests in full on the earlier of the first anniversary of the date of grant or the date immediately prior to our next annual meeting of stockholders. |
Director Compensation Paid for 2016
The following table summarizes all compensation paid to or earned by our non-employee directors as compensation for board service during the 2016 fiscal year.
|
Name |
|
Fees Earned or Paid in Cash |
|
Option Awards |
|
|
Total |
|
Neil Clendeninn |
|
60,000 |
|
14,012 |
|
|
74,012 |
|
Jack Goldstein |
|
87,500 |
|
19,617 |
|
|
107,117 |
|
Martin Mattingly |
|
57,500 |
|
14,012 |
|
|
71,512 |
|
Stewart Parker |
|
60,000 |
|
14,012 |
|
|
74,012 |
|
David Smith |
|
65,000 |
|
14,012 |
|
|
79,012 |
|
|
|
|
|
|
|
|
|
|
(1) |
The dollar amounts reflect the aggregate grant date fair value of equity awards granted within the fiscal year in accordance with the Financial Accounting Standards Board, or FASB, Accounting Standards Codification Topic 718 for stock-based compensation. These amounts do not correspond to the actual cash value that will be recognized by the directors when received. Assumptions used in the calculation of the amounts in this column are included in note 10 to our audited consolidated financial statements included in this 2016 Annual Report on Form 10-K. As of December 31, 2016, the following directors had the following number of options outstanding: |
|
|
• |
Neil Clendeninn: 48,500 options, of which 26,000 were vested as of December 31, 2016. |
|
|
• |
Jack Goldstein: 59,461 options, of which 27,961 were vested as of December 31, 2016. |
|
|
• |
Martin Mattingly: 50,500 options, of which 28,000 were vested as of December 31, 2016. |
|
|
• |
Stewart Parker: 50,461 options, of which 27,961 were vested as of December 31, 2016. |
|
|
• |
David Smith: 50,500 options, of which 28,000 were vested as of December 31, 2016. |
|
(2) |
These options were granted on May 26, 2016 under our 2010 Performance Incentive Plan and vest 100% on the earlier of the one-year anniversary of the date of grant or the day immediately prior to the 2017 Annual Meeting of Stockholders. |
103
The following table sets forth certain information regarding our equity compensation plans as of December 31, 2016:
|
|
|
(a) |
|
|
(b) |
|
|
(c) |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
Number of securities |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
remaining available for |
|
|
|
|
|
|
Number of securities to be |
|
|
|
|
|
|
future issuance under |
|
|
||
|
|
|
issued upon exercise of |
|
|
Weighted-average |
|
|
equity compensation |
|
|
|||
|
|
|
outstanding options, |
|
|
exercise price of |
|
|
plans (excluding |
|
|
|||
|
|
|
restricted stock units, |
|
|
outstanding options, |
|
|
securities reflected in |
|
|
|||
|
Plan category |
|
warrants and rights |
|
|
warrants and rights |
|
|
column (a)) |
|
|
|||
|
Equity compensation plans approved by security holders |
|
|
1,632,026 |
|
(1) |
$ |
7.28 |
|
(1) |
|
2,002,032 |
|
(1) |
|
Equity compensation plans not approved by security holders(2) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
Total |
|
|
1,632,026 |
|
|
$ |
7.28 |
|
|
|
2,002,032 |
|
|
|
(1) |
As of December 31, 2016, we maintained the following equity compensation plans, which were approved by security holders: (a) the 2000 Stock Incentive Plan, (b) the 2007 Performance Incentive Plan, (c) the OncoGenex Technologies Amended and Restated Stock Option Plan and (d) the 2010 Performance Incentive Plan. |
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth certain information with respect to the beneficial ownership of our common stock by the following persons as of February 14, 2017, except as otherwise noted in the footnotes to the table:
|
|
• |
each person, entity or group who we know to beneficially own five percent or more of our voting securities; |
|
|
• |
each of our directors and director nominees; |
|
|
• |
each of our Named Executive Officers identified in the Summary Compensation Table; and |
|
|
• |
all of our directors and executive officers as a group. |
104
The address of each beneficial owner listed in the table is c/o OncoGenex Pharmaceuticals, Inc., 19820 North Creek Parkway, Suite 201, Bothell, Washington 98011. The percentages in the table below are based on 30,025,521 shares of our common stock outstanding as of December 31, 2016. Except as indicated in the footnotes to the table and pursuant to applicable community property laws, to our knowledge, each stockholder named in the table has sole voting and investment power with respect to the shares set forth opposite such stockholder’s name. The information provided in the table is based on our records and information filed with the SEC, unless otherwise noted.
|
Name of Beneficial Owner |
Amount and Nature of |
Percent of Class(%)(1) |
|
Named Executive Officers and Directors: |
|
|
|
Scott Cormack(2) |
577,599 |
1.9 |
|
Cindy Jacobs(3) |
191,461 |
* |
|
John Bencich (4) |
50,863 |
* |
|
Neil Clendeninn(5) |
48,421 |
* |
|
Stewart Parker(6) |
40,461 |
* |
|
Jack Goldstein(7) |
45,461 |
* |
|
Martin Mattingly(8) |
38,500 |
* |
|
David Smith(9) |
37,500 |
* |
|
All current officers and directors as a group (8 persons) (10) |
1,030,266 |
3.4 |
|
* Less than 1% |
|
|
|
(1) |
Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. Shares of common stock subject to options and warrants currently exercisable, or exercisable within 60 days of February 14, 2017, are deemed outstanding for computing the percentage of the person holding such options or warrants but are not deemed outstanding for computing the percentage of any other person. |
|
(2) |
Represents 140,050 shares owned directly, 220,205 options, 84,055 shares owned indirectly through his spouse and 133,289 options owned indirectly through his spouse exercisable within 60 days of February 14, 2017. |
|
(3) |
Represents 84,359 shares owned directly and 107,102 options exercisable within 60 days of February 14, 2017. |
|
(4) |
Represents 11,488 shares owned directly and 39,375 options exercisable within 60 days of February 14, 2017. |
|
(5) |
Represents 22,421 shares owned directly and 26,000 options exercisable within 60 days of February 14, 2017. |
|
(6) |
Represents 12,500 shares owned directly and 27,961 options exercisable within 60 days of February 14, 2017. |
|
(7) |
Represents 17,500 shares owned directly and 27,961 options exercisable within 60 days of February 14, 2017. |
|
(8) |
Represents 10,500 shares owned directly and 28,000 options exercisable within 60 days of February 14, 2017. |
|
(9) |
Represents 9,500 shares owned directly and 28,000 options exercisable within 60 days of February 14, 2017. |
|
(10) |
Represents for the current officers and directors as a group, 392,373 shares owned directly or indirectly as indicated above, and 637,893 options exercisable within 60 days of February 14, 2017. |
Related-Party Transactions Policy and Procedure
Our Audit Committee is responsible for reviewing and approving all related-party transactions and conflict of interest situations involving a principal stockholder, a member of the Board of Directors or senior management. Our Code of Conduct and Business Ethics requires our executive officers and directors to report any conflicts of interest with our interests to our Audit Committee, and generally prohibits our executive officers and directors from conflicts of interest with our interests. Waivers of our Code of Conduct and Business Ethics with respect to an executive officer or director may only be granted by the Board of Directors or, if permitted by NASDAQ and any other applicable stock exchange’s rules, our Nominating and Governance Committee. We do not have a specific policy concerning approval of transactions with stockholders who own more than five percent of our outstanding shares.
Other than as disclosed below and in “Compensation Discussion and Analysis” and “Executive Compensation,” from January 1, 2016 to the present, there have been no transactions, and there are currently no proposed transactions, in which the amount involved exceeds $120,000 to which we or any of our subsidiaries was or is to be a party and in which any director, director nominee, executive officer, holder of more than 5% of our capital stock, or any immediate family member of any of these individuals, had or will have a direct or indirect material interest.
In January 2016, Scott Cormack, our Chief Executive Officer, married Michelle Griffin, a consultant to our company. Pursuant to a consulting agreement entered into April 1, 2013 and updated periodically with revised statements of work, during 2016, we paid Ms.
105
Griffin approximately $0.5 million for consulting services. We also granted Ms. Griffin performance-based options to purchase 135,000 shares of common stock in 2016. The performance milestones pursuant to which the options would have vested were not achieved and as of December 31, 2016, the options were cancelled.
Director Independence
Our Board of Directors has determined that each of our nominees for director, other than Mr. Cormack, is “independent” under the applicable Securities and Exchange Commission, or the SEC, rules and the criteria established by The Nasdaq Stock Market LLC, or NASDAQ.
Our Board of Directors has also determined that each member of our Audit Committee, Compensation Committee and Nominating and Governance Committee meets the independence standards applicable to those committees prescribed by the applicable rules and regulations of NASDAQ and the SEC.
Fees Billed by Independent Registered Public Accounting Firm
The following is a summary of the fees billed by our independent registered public accounting firm for the fiscal years ended December 31, 2016 and December 31, 2015 for professional services rendered to us:
|
Fee Category |
Fiscal 2016 Fees(1) |
Fiscal 2015 Fees(1) |
|
Audit Fees |
$289,463(2) |
$332,728(2) |
|
Audit-Related Fees |
- |
- |
|
Tax Fees |
- |
- |
|
All Other Fees |
- |
- |
|
Total Fees |
$289,463 |
$332,728 |
|
(1) |
Accountant fees and services charged by Ernst & Young LLP are paid in Canadian dollars and shown in USD. For fiscal 2016, the fees were CDN$289,463 and was converted at an average exchange rate of US$1.00 = CDN$1.3333. For fiscal 2015, the fees were CDN$332,728 and was converted at an average exchange rate of US$1.00 = CDN$1.3333. |
|
(2) |
Audit Fees for 2016 and 2015 are fees billed and to be billed for the audit of our consolidated financial statements, review of the consolidated financial statements included in our quarterly reports, and for services in connection with regulatory filings and engagements. |
Audit Fees. Consists of fees billed for professional services rendered for the audit of our consolidated financial statements and review of the interim consolidated financial statements included in quarterly reports on Form 10-Q that are filed with the SEC.
Audit-Related Fees. Consists of fees billed for assurance and related services that are reasonably related to the performance of the audit or review of our consolidated financial statements, including accounting consultations and fees related to registration of securities.
Policy on Audit Committee Pre-Approval of Audit Services and Permissible Non-Audit Services
The Audit Committee’s policy is to pre-approve all audit and permissible non-audit services performed by our independent registered public accounting firm. These services may include audit services, audit-related services, tax services and other services. For audit services, our independent registered public accounting firm typically provides audit service detail in advance of the second quarter meeting of the Audit Committee, which outlines the scope of the audit and related audit fees. If agreed to by the Audit Committee, an engagement letter is formally accepted by the Audit Committee.
For non-audit services, our senior management will submit from time to time to the Audit Committee for approval non-audit services that it recommends the Audit Committee engage our independent registered public accounting firm to provide for the fiscal year. Our senior management and our independent registered public accounting firm will each confirm to the Audit Committee that each non-audit service is permissible under all applicable legal requirements. A budget, estimating non-audit service spending for the fiscal year, will be provided to the Audit Committee along with the request. The Audit Committee must approve both permissible non-audit services and the budget for such services. The Audit Committee will be informed routinely as to the non-audit services actually provided by our independent registered public accounting firm pursuant to this pre-approval process.
For the 2015 and 2016 fiscal years, the Audit Committee approved all of the services provided by Ernst & Young LLP described above.
106
(1) Financial Statements
(2) All schedules are omitted because they are not required or the required information is included in the consolidated financial statements or notes thereto.
(3) Exhibits
|
Exhibit |
|
Description |
|
Incorporated by Reference |
|
Filed/ |
||||||
|
|
|
Form |
|
File No. |
|
Exhibit |
|
Filing Date |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.1† |
|
Agreement and Plan of Merger and Reorganization, dated as of January 5, 2017, by and among OncoGenex Pharmaceuticals, Inc., Ash Acquisition Sub, Inc., Ash Acquisition Sub 2, Inc. and Achieve Life Science, Inc. |
|
8-K |
|
033-80623 |
|
2.1 |
|
January 5, 2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3.1 |
|
Second Amended and Restated Certificate of Incorporation filed on May 24, 2013 |
|
8-K |
|
033-80623 |
|
3.1 |
|
May 29, 2013 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3.2 |
|
Certificate of Amendment to Amended and Restated Certificate of Incorporation |
|
8-K |
|
033-80623 |
|
3.1 |
|
May 22, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3.3 |
|
Sixth Amended and Restated Bylaws of OncoGenex Pharmaceuticals, Inc. |
|
8-K |
|
033-80623 |
|
3.1 |
|
January 5, 2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.1 |
|
Specimen Certificate of Common Stock |
|
10-Q |
|
000-21243 |
|
4.1 |
|
November 10, 2008 |
|
|
|
4.7 |
|
Form of Series A Warrant |
|
8-K |
|
033-80623 |
|
4.1 |
|
June 27, 2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.8 |
|
Form of Series A-1 Warrant |
|
8-K |
|
033-80623 |
|
4.1 |
|
April 30, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.9 |
|
Form of Pre-Funded Series B Warrant |
|
8-K |
|
033-80623 |
|
4.2 |
|
June 27, 2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.10 |
|
Form of Series B Warrant |
|
8-K |
|
033-80623 |
|
4.3 |
|
June 27, 2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1 |
|
Sonus Pharmaceuticals, Inc. 2000 Stock Incentive Plan (the “2000 Plan”)†† |
|
10-Q |
|
000-21243 |
|
10.41 |
|
August 14, 2000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.2 |
|
First Amendment to Sonus Pharmaceuticals, Inc. 2000 Plan†† |
|
10-Q |
|
000-21243 |
|
10.23 |
|
November 8, 2006 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.3 |
|
Form of Sonus Pharmaceuticals, Inc. Stock Option Agreement (pertaining to the 2000 Plan)†† |
|
10-Q |
|
000-21243 |
|
10.42 |
|
August 14, 2000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.4 |
|
Sonus Pharmaceuticals, Inc. 2007 Performance Incentive Plan (the “2007 Plan”)†† |
|
DEF 14A |
|
000-21243 |
|
Appendix A |
|
April 3, 2007 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
107
108
|
Exhibit |
|
Description |
|
Incorporated by Reference |
|
Filed/ |
||||||
|
|
|
Form |
|
File No. |
|
Exhibit |
|
Filing Date |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
License Agreement between OncoGenex Technologies Inc. and the University of British Columbia effective as of April 5, 2005, and Amending Agreement dated as of August 30, 2006 (OGX-427)* |
|
F-1, Amendment No. 1 |
|
333-139293 |
|
10.14 |
|
January 29, 2007 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.23 |
|
Second Amending Agreement as of August 7, 2008 between the University of British Columbia and OncoGenex Technologies Inc. (OGX-427) |
|
10-Q |
|
000-21243 |
|
10.40 |
|
November 10, 2008 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.24 |
|
Office Lease by and between Grosvenor International (Atlantic Freeholds) Limited and OncoGenex Pharmaceuticals, Inc., dated February 11, 2015 |
|
8-K |
|
033-80623 |
|
10.1 |
|
February 12, 2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.25 |
|
Form of Support Agreement, by and between OncoGenex Pharmaceuticals, Inc. and certain directors, officers and stockholders of Achieve Life Science, Inc. |
|
8-K |
|
033-80623 |
|
10.1 |
|
January 5, 2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.26 |
|
Form of Support Agreement, by and between Achieve Life Science, Inc. and certain directors and officers of OncoGenex Pharmaceuticals, Inc. |
|
8-K |
|
033-80623 |
|
10.2 |
|
January 5, 2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.27 |
|
Form of Lock-Up Agreement, by and between OncoGenex Pharmaceuticals, Inc. and certain directors, officers and stockholders of Achieve Life Science, Inc. and OncoGenex Pharmaceuticals, Inc. |
|
8-K |
|
033-80623 |
|
10.3 |
|
January 5, 2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.28 |
|
Form of CVR Agreement, by and between OncoGenex Pharmaceuticals, Inc., Achieve Life Science, Inc. and a Rights Agent to be determined. |
|
8-K |
|
033-80623 |
|
10.4 |
|
January 5, 2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
21.1 |
|
Subsidiaries of the Registrant |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
23.1 |
|
Consent of Ernst & Young LLP |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
24.1 |
|
Power of Attorney (included on the signature page hereto) |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.1 |
|
Certification of Chief Executive pursuant to Rule 13a-14(a) or 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.2 |
|
Certification of Chief Financial Officer pursuant to Rule 13a-14(a) or 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.1 |
|
Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002** |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
109
|
Exhibit |
|
Description |
|
Incorporated by Reference |
|
Filed/ |
||||||
|
|
|
Form |
|
File No. |
|
Exhibit |
|
Filing Date |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Certification of Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002** |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.INS |
|
XBRL Instance Document |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.SCH |
|
XBRL Taxonomy Extension Schema Document |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.CAL |
|
XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
|
|
|
|
|
|
X |
|
101.DEF |
|
XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.LAB |
|
XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.PRE |
|
XBRL Taxonomy Extension Presentation Linkbase Document |
|
|
|
|
|
|
|
|
|
X |
|
† |
Schedules and similar attachments to the Arrangement Agreement have been omitted pursuant to Item 601(b)(2) of Regulation S-K. The Company will furnish supplementally a copy of any omitted schedule or similar attachment to the SEC upon request. |
|
†† |
Indicates management contract or compensatory plan or arrangement. |
|
* |
Confidential portions of this exhibit have been omitted and filed separately with the Commission pursuant to an application for Confidential Treatment under Rule 24b-2 promulgated under the Securities Exchange Act of 1934, as amended. |
|
** |
The certifications attached as Exhibits 32.1 and 32.2 accompany to this Annual Report on Form 10-K pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, and shall not be deemed filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended. |
110
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
|
|
|
ONCOGENEX PHARMACEUTICALS, INC. |
||
|
|
|
|
|
(Registrant) |
||
|
|
|
|
|
|||
|
Date: February 23, 2017 |
|
|
|
By: |
|
/s/ SCOTT CORMACK |
|
|
|
|
|
|
|
Scott Cormack |
|
|
|
|
|
|
|
President and Chief Executive Officer |
Power of Attorney
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Scott Cormack and John Bencich, jointly and severally, as such person’s attorneys-in-fact, each with the power of substitution, for such person in any and all capacities, to sign any amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
By: /s/ SCOTT CORMACK |
|
President and Chief Executive Officer |
|
Date: February 23, 2017 |
|
Scott Cormack |
|
|
|
|
|
|
|
|
||
|
By: /s/ JOHN BENCICH |
|
Chief Financial Officer |
|
Date: February 23, 2017 |
|
John Bencich |
|
|
|
|
|
|
|
|
||
|
By: /s/ JACK GOLDSTEIN |
|
Chairman of the Board and Director |
|
Date: February 23, 2017 |
|
Jack Goldstein |
|
|
|
|
|
|
|
|
||
|
By: /s/ NEIL CLENDENINN |
|
Director |
|
Date: February 23, 2017 |
|
Neil Clendeninn |
|
|
|
|
|
|
|
|
||
|
By: /s/ MARTIN MATTINGLY |
|
Director |
|
Date: February 23, 2017 |
|
Martin Mattingly |
|
|
|
|
|
|
|
|
||
|
By: /s/ H. STEWART PARKER |
|
Director |
|
Date: February 23, 2017 |
|
H. Stewart Parker |
|
|
|
|
|
|
|
|
||
|
By: /s/ DAVID SMITH |
|
Director |
|
Date: February 23, 2017 |
|
David Smith |
|
|
|
|
111