10-K: Annual report pursuant to Section 13 and 15(d)
Published on March 16, 2007
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
x ANNUAL
REPORT PURSUANT TO SECTION 13 OR 15 (d) OF
THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2006
Or
o TRANSITION
REPORT PURSUANT TO SECTION 13 OR 15 (d) OF THE
SECURITIES AND EXCHANGE ACT OF 1934 (NO FEE REQUIRED)
Commission File Number 0-26866
Sonus Pharmaceuticals, Inc.
(Exact name of the registrant as specified in its charter)
|
Delaware |
|
95-4343413 |
|
(State or other jurisdiction of |
|
(I.R.S. Employer |
|
incorporation or organization) |
|
Identification No.) |
22026 20th Avenue SE, Bothell, Washington 98021
(Address of principal executive offices)
(425) 487-9500
(Registrants telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of Each Class |
|
Name of Exchange on Which Registered |
|
Common Stock, par value $0.001 per share |
|
The NASDAQ Stock Market, LLC |
|
Series A
Junior Participating Preferred Stock, |
|
The NASDAQ Stock Market, LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 of 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months ( or for such shorter period that the registrant was required to file such report), and (2) has been subject to such filing requirements for the past 90 days. Yes o No x
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrants knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of accelerated filer and large accelerated filer in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer o |
|
Accelerated filer x |
|
Non-accelerated filer o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act.). Yes o No x
As of June 30, 2006, the aggregate market value of the registrants Common Stock held by non-affiliates of the registrant was $162,703,119. As of March 1, 2007, 36,853,974 shares of the registrants Common Stock were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrants definitive Proxy Statement to be filed in connection with the solicitation of proxies for its 2007 Annual Meeting of Stockholders to be held on May 10, 2007 are incorporated by reference in Items 10, 11, 12, 13 and 14 of Part III hereof.
Sonus Pharmaceuticals, Inc.
Table of Contents
2
References in this Form 10-K to Sonus Pharmaceuticals, Sonus, the Company, we, us or our refer to Sonus Pharmaceuticals, Inc. The information in this Form 10-K contains certain forward-looking statements, including statements related to clinical trials, regulatory approvals, markets for the Companys products, new product development, capital requirements and trends in its business that involve risks and uncertainties. The Companys actual results may differ materially from the results discussed in the forward-looking statements. Factors that might cause such a difference include those discussed in Business, Certain Factors that May Affect Our Business and Future Results and Managements Discussion and Analysis of Financial Condition and Results of Operations as well as those discussed elsewhere in this Form 10-K.
Overview
Sonus Pharmaceuticals is focused on the development of cancer drugs that are designed to provide better efficacy, safety and tolerability, and are easier to use. Our business strategy is as follows:
· develop proprietary formulations of cancer drugs utilizing our TOCOSOL® technology;
· develop novel formulations of oncology related drugs; and
· identify and acquire products/technologies that are complementary to our focus in oncology in order to broaden our business and market opportunities.
Proprietary TOCOSOL technology
Our novel vitamin E-based emulsion technology has been designed to address the challenges of poorly soluble cancer drugs. The technology uses vitamin E oil and tocopherol derivatives to solubilize and formulate drugs with the goal of enhancing their efficacy, safety and administration. Drug products formulated with our TOCOSOL technology are ready-to-use, requiring no dilution or reconstitution.
Our lead oncology product candidate, TOCOSOL Paclitaxel, is a novel, nanodroplet formulation of paclitaxel, one of the worlds most widely prescribed anti-cancer drugs. Paclitaxel, a member of the taxane family of cancer drugs, is the active ingredient in Taxol®, which is approved in the U.S. for the treatment of breast, ovarian and non-small cell lung cancers and Kaposis sarcoma. Our product, TOCOSOL Paclitaxel, is a ready-to-use, injectable paclitaxel emulsion formulation. We believe that data from our Phase 2 clinical trials conducted to-date suggest that TOCOSOL Paclitaxel:
· eliminates the need for cremophor, which is used in Taxol and generic paclitaxel and has known toxicities;
· compares favorably with approved taxane products and other new paclitaxel formulations under development (safety and efficacy remain to be proven in Phase 3 testing of TOCOSOL Paclitaxel, which is currently underway);
· offers the convenience of a ready-to-use formulation that does not require preparation prior to administration;
· can be administered to patients by a short 15-minute infusion, compared to the one- to three-hour infusion that is typically required with Taxotere® and Taxol or generic versions of paclitaxel;
· does not require any special intravenous (i.v.) tubing or filters; and
· can be administered in small volumes of 15 to 35 milliliters compared to volumes of several hundred milliliters of i.v. solution that are required for dosing of Taxol or Taxotere.
We initiated Phase 2a studies for TOCOSOL Paclitaxel in March 2002 to evaluate the safety and efficacy of TOCOSOL Paclitaxel in ovarian, non-small cell lung and bladder cancers using weekly dosing of the product. These were single agent, open label studies that enrolled taxane naïve patients who had progressive disease despite prior treatment with a standard chemotherapy regimen. The best tolerated dose of TOCOSOL Paclitaxel administered weekly was initially estimated to be 120 mg/m² per week in the ovarian and lung cancer trials, and 100 mg/m² per week in the bladder cancer trial, based on observations among a small number of patients treated for a few weeks. Subsequent review of actual doses administered across all patients in all studies over extended treatment periods indicated that patients assigned to receive weekly doses of
3
100 mg/m² or 120 mg/m² actually received similar cumulative doses over time, based on long-term tolerability. Patient enrollment in the Phase 2a clinical trials was completed in the second quarter of 2003. Data review, confirmation and analysis are now complete, and databases have been locked. A total of 120 patients in the ovarian, non-small cell lung and bladder cancer studies were evaluable for objective response, which means that the patients received at least eight weekly cycles of TOCOSOL Paclitaxel and underwent CT scans to determine anti-tumor responses according to RECIST. Patients were also evaluated for time to disease progression and overall survival. Final analyses of all data are now complete and response rates are presented in the table below.
|
|
|
No. |
|
|
|
Objective Responses (OR) |
|
||||||||
|
Cancer Type |
|
Patients |
|
Stable |
|
Partial |
|
Complete |
|
Total |
|
% |
|
95% |
|
|
Ovarian |
|
52 |
|
15 |
|
17 |
|
3 |
|
20 |
|
38 |
% |
(25% - 53%) |
|
|
NSCL |
|
43 |
|
19 |
|
6 |
|
3 |
|
9 |
|
21 |
% |
(10% - 36%) |
|
|
Bladder |
|
27 |
|
11 |
|
7 |
|
2 |
|
9 |
|
33 |
% |
(17% - 54%) |
|
Following completion of treatment in the Phase 2a studies, clinical monitoring of each consenting patient was continued to assess survival duration. Median survival in each of the three studies has been estimated based on reports received from investigators:
|
Cancer Type |
|
Median |
|
95% CI |
|
|
Ovarian |
|
64.1 |
|
(49.1 - 106.4) |
|
|
NSCL |
|
34.7 |
|
(18.9 - 48.0) |
|
|
Bladder |
|
57.4 |
|
(27.1 - 94.9) |
|
In September 2004, we initiated a Phase 2b study of TOCOSOL Paclitaxel for first line treatment of women with metastatic breast cancer. Enrollment in this study was closed in October 2004 with 47 patients randomized. The investigators reported an overall objective response rate of 53%, (95% Confidence Interval 38% - 68%). Review of all radiographic images by an independent radiologist who had no information about individual patients treatment or non-radiographic response assessments reported a confirmed objective response rate of 49%, (95% Confidence Interval 34% - 64%).
In addition to being assessed for anti-tumor efficacy, patients are also monitored for adverse events in all clinical studies. The most significant adverse events expected with taxanes are neutropenia and peripheral neuropathy. Among 232 patients treated in the Phase 2 clinical trials, the incidence of at least one episode of Grade 4 neutropenia (absolute neutrophil count <500 cells/mm3) during treatment was 18%. However, only 2% of patients had febrile neutropenia, and there was one septic death. No peripheral neuropathy was observed in 56% of patients, Grade 3 peripheral neuropathy was reported in only 10% of patients cumulatively, and no patients experienced Grade 4 peripheral neuropathy. We believe these adverse event rates compare favorably to the reported neutropenia and peripheral neuropathy experienced when Taxol is administered with the approved dosing regimen of 175 mg/m2 every three weeks. Dose reductions or treatment delays due to toxicity from TOCOSOL Paclitaxel did not limit long-term treatment in most patients. A majority of patients in our Phase 2 studies were administered antihistamines prior to the infusion of TOCOSOL Paclitaxel. Paclitaxel-mediated infusion-related toxicities, sometimes called hypersensitivity reactions were generally mild and were reported following approximately 11% of all doses. Investigators have reported that infusion-related toxicities associated with our product could be ameliorated by temporary (a few minutes) interruption of infusion and restarting the infusion at a slower rate. Infusion-related toxicities very rarely prevented delivery of intended doses. Overall, we believe that TOCOSOL Paclitaxel appears to be well tolerated over multiple treatment cycles.
The results of the Phase 2 clinical trials may or may not be indicative of the final results upon completion of the ongoing studies or our Phase 3 pivotal study that was initiated in September 2005 and closed enrollment in November 2006.
The manufacturing process for TOCOSOL Paclitaxel has been successfully scaled to support commercialization. In March 2005, Sonus met with the U.S. Food and Drug Administration (FDA) to discuss the Chemistry, Manufacturing and Controls (CMC) data for TOCOSOL Paclitaxel. The FDA did not identify any issues with the manufacture and control of the drug product that would preclude Sonus from using TOCOSOL Paclitaxel in the Phase 3 trial, nor from Bayer Schering Pharma AG (Bayer Schering) from submitting the intended New Drug Application (NDA) based on the results of that trial.
4
Our objective is to work with our corporate partner (Bayer Schering) to advance final clinical development, gain marketing approval and maximize the commercial opportunity for TOCOSOL Paclitaxel. Our regulatory strategy is to gain the fastest possible market entry with a competitive label, while pursuing opportunities to further differentiate the product. Our strategy for product approval includes the following:
· U.S. Development. In collaboration with our partner, we will seek initial approval of TOCOSOL Paclitaxel with a 505(b)(2) NDA submission, which will rely on the FDAs previous findings of safety and efficacy for Taxol (the reference paclitaxel product), supplemented by data supporting TOCOSOL Paclitaxels safety and efficacy. After meetings with the FDA, and based on preclinical and clinical data generated to date, the FDA indicated that it was appropriate for Sonus to conduct a single Phase 3 clinical trial that would be the basis for submission of a NDA for TOCOSOL Paclitaxel under the 505(b)(2) regulatory mechanism. The FDA and Sonus finalized the study design and plans for conducting and analyzing the results of the Phase 3 trial under a Special Protocol Assessment (SPA), which was completed in June 2005. The Phase 3 study is comparing the safety and efficacy of TOCOSOL Paclitaxel administered weekly with Taxol administered weekly. Enrollment in the Phase 3 study was closed in November 2006 with 821 patients randomized.
The FDA has indicated to Sonus that a NDA approval will require either (a) demonstration of superior efficacy of TOCOSOL Paclitaxel compared to Taxol; or (b) demonstration of non-inferior efficacy as compared to Taxol and either (i) a change of the approved label for Taxol to include a weekly dosing schedule or (ii) availability of reviewable data from a trial demonstrating superior efficacy of Taxol using a weekly dosing schedule as compared to that of Taxol using the currently approved three-weekly dosing schedule.
We have an agreement with the Cancer and Leukemia Group B Foundation (the CALGB) giving us the right to use data from the CALGB Study 9840, a Phase 3 trial comparing weekly dosing of Taxol to three-weekly dosing of Taxol in patients with metastatic breast cancer. In the event that TOCOSOL Paclitaxel does not achieve superior efficacy over Taxol in the Phase 3 trial or the approved label for Taxol is not changed to include a weekly dosing schedule, Bayer Schering and we plan to submit the CALGB 9840 data to the FDA as part of the TOCOSOL Paclitaxel NDA to support weekly dosing of Taxol as an appropriate reference arm in the ongoing pivotal Phase 3 trial. Based on the summary presentation of CALGB 9840 at the American Society of Clinical Oncology (ASCO) 2004 annual meeting, our analysis of the data set and discussions with the FDA, we believe that the data from this study should fulfill the FDAs requirement to submit a reviewable data set that compares weekly dosing of Taxol to three-weekly dosing of Taxol, and demonstrate the weekly regimen to be superior to the every three-weekly regimen. While there can be no assurance that the data obtained from this study will be sufficient to support the TOCOSOL Paclitaxel NDA, Sonus preliminary analysis of the CALGB 9840 data confirm the conclusions presented in the abstract at ASCO 2004. If the FDA does not accept the CALGB Study 9840 data as support for a weekly reference arm, substantial additional costs and time would be required before the NDA submission for TOCOSOL Paclitaxel.
The clinical trial protocol and Statistical Analysis Plan approved under the SPA provide for sequential superiority analyses for efficacy of TOCOSOL Paclitaxel compared to Taxol, provided that we first demonstrate a non-inferior objective response rate; however, there can be no assurance that the Phase 3 clinical trial data will demonstrate that TOCOSOL Paclitaxel has efficacy that is non-inferior or superior to Taxol.
We are collaborating with Bayer Schering on the assembly of the NDA and are completing those components which are the responsibility of Sonus within timelines established by Bayer Schering. Bayer Schering is responsible for submission of the NDA, which we currently believe will be in the second quarter of 2008. However, this is only an estimate as the submission date is outside of our direct control and is subject to change.
· Ex-U.S. development. Our corporate partner will pursue development and approval of TOCOSOL Paclitaxel in markets outside the U.S. (initially Europe and Japan). During the second quarter of 2006, Bayer Scherings subsidiary in Japan submitted the Japanese Investigational New Drug Application (IND) for TOCOSOL Paclitaxel, which was accepted by the regulatory authorities. A Phase 1 study was initiated in the third quarter of 2006 subsequent to the acceptance of the IND, the results of which will also be used to support U.S. regulatory submissions.
· New indications for taxanes. In conjunction with our corporate partner, we may pursue clinical development of TOCOSOL Paclitaxel for the treatment of other types of cancer, including indications for which Taxol has been approved as well as for diseases for which Taxol is used but not approved. In October 2003, we announced that we were granted Fast Track designation by the FDA for the development of TOCOSOL Paclitaxel for inoperable or metastatic urothelial transitional cell cancers (mostly urinary bladder cancers). In December 2004, the FDA granted an Orphan Drug designation to TOCOSOL Paclitaxel for the treatment of non-superficial urothelial
5
cancer. We initiated a Phase 2b study in bladder cancer in the U.S. during the fourth quarter of 2003, and in Spain and the U.K. during 2005, using weekly dosing of TOCOSOL Paclitaxel. Enrollment in this trial was completed in September 2006 and we expect to have preliminary data by mid-2007. Continued development in this indication will be dependent on many factors, including the clinical and commercial potential compared to other opportunities in our pipeline and agreement with Bayer Schering to jointly develop.
The scope, timing and costs of the clinical trials to be conducted under all of the above regulatory strategies are difficult to determine with accuracy. We are conducting a single pivotal Phase 3 trial in metastatic breast cancer, an indication where paclitaxel is approved, with a primary endpoint of objective response rate and secondary endpoints of progression-free survival and overall survival durations. Our partner expects to submit the NDA with data on the primary endpoint, potentially followed by supplemental submissions for the secondary endpoints when data are mature. The Phase 3 trial, which compares TOCOSOL Paclitaxel to Taxol administered weekly, is powered to achieve statistical significance on all three endpoints, and was designed to enroll approximately 800 evaluable patients. Our current estimate for the total cost of the pivotal Phase 3 trial is approximately $50 million. This estimate is our external direct cost only and does not include any internal costs or future billings from Bayer Schering. Under our Collaboration and License Agreement with Bayer Schering, dated October 17, 2005, Bayer Schering will fund 50% of these costs (in certain cases the reimbursement rate is 100%). In addition, it is anticipated that we will collaborate with Bayer Schering on additional studies of TOCOSOL Paclitaxel. Under the terms of our agreement, we are obligated to fund 50% of the costs of any additional studies conducted by Bayer Schering in support of commercialization activities for the U.S. market. The exact cost and timing of these studies is yet to be finalized. The current ongoing Phase 3 trial will constitute the bulk of the Companys clinical trial spending in 2007. Approximately two thirds of the cost of the Phase 3 trial has been incurred as of December 31, 2006. However, future costs may vary significantly depending upon regulatory and other matters that are not within our control and there can be no assurance that such amount will be sufficient to complete the study. Development costs for commercialization activities outside the U.S. market are borne by Bayer Schering. There can be no assurance that the results of any or all of the anticipated clinical trials will be successful or will support product approval.
Our second oncology drug candidate is TOCOSOL Camptothecin Injectable Emulsion. This product candidate is a novel camptothecin derivative formulated as an oil-in-water emulsion with Sonus proprietary TOCOSOL technology. Camptothecins are an important class of anti-cancer drugs introduced in recent years; however, the marketed camptothecin analogs, irinotecan (Camptosar®) and topotecan (Hycamtin®), have demonstrated limitations that may reduce their clinical utility. Irinotecan and topotecan are used in the treatment of colorectal, lung, ovarian and cervical cancers. The active ingredient in TOCOSOL Camptothecin is SN-38 (formulated as a prodrug), which is the active ingredient in irinotecan. Our objective with TOCOSOL Camptothecin is to provide a ready to use product that has enhanced anti-tumor activity and improved tolerability compared with the approved camptothecin-based products. An IND was submitted to the FDA for TOCOSOL Camptothecin in June 2006. The FDA completed its review in July 2006, and Phase 1 clinical testing was initiated in September 2006. Our research and development efforts on TOCOSOL Camptothecin enabled the initiation of the Phase 1 study; however, we cannot give any assurance that this compound will be clinically successful.
Research and Development Pipeline
We continue to invest in the research and development of new oncology related product candidates, including those that we believe could extend the application of our technology. In addition to our internal research and development efforts, we may also consider acquisitions of other products, development candidates or technologies to expand our pipeline and capabilities.
Market Overview
Cancer is characterized by rapid, uncontrolled cell division resulting in the growth of an abnormal mass of cells generally referred to as a malignant tumor. Cancerous tumors can arise in almost any tissue or organ, and cancer cells, if not eradicated, can spread, or metastasize, throughout the body. As these tumors grow, they cause damage to the surrounding tissue and organs and commonly result in death if left untreated. Cancer is believed to occur as a result of a number of hereditary and environmental factors. According to the American Cancer Society, cancer is the second leading cause of death in the United States and accounts for approximately one in every four deaths. Approximately 560,000 Americans are expected to die of cancer in 2007. The National Institutes of Health estimated the direct medical cost of cancer to be $78 billion in 2006.
Our lead product candidate, TOCOSOL Paclitaxel, is a chemotherapy drug incorporating paclitaxel as the active ingredient. If approved, TOCOSOL Paclitaxel will be a member of the taxane class of chemotherapy drugs, which generates annual worldwide sales estimated to be in excess of $3.5 billion. TOCOSOL Paclitaxel would address a portion of this
6
market depending on the approved indication(s). Other product candidates in our pipeline are in the early stages of development, and it is difficult to evaluate the potential markets for these product candidates as the areas of potential application are diverse and specific applications are yet to be determined.
Manufacturing
We originally used the University of Iowa as the FDA-approved institution to manufacture TOCOSOL Paclitaxel under current Good Manufacturing Practice (GMP) requirements for our use in preclinical and clinical studies. In mid-2002, we entered into a manufacturing and supply agreement with Sicor Pharmaceutical Sales, Inc. (Sicor is now known as TEVA Pharmaceuticals USA). During 2003, in collaboration with TEVA Pharmaceuticals USA, we completed scale-up of the drug product manufacturing process for TOCOSOL Paclitaxel to commercial scale under current GMP requirements, ensuring adequate clinical drug supplies for ongoing and planned clinical trials, and providing a commercial process to enable regulatory approval and commercial product launch.
On March 2, 2006, in accordance with the Collaboration and License Agreement with Bayer Schering, Bayer Schering exercised their right to assume responsibility for the manufacturing of TOCOSOL Paclitaxel. In June 2006, we entered into a clinical supply agreement with Bayer Schering to provide clinical supplies of TOCOSOL Paclitaxel to Bayer Schering until such time as Bayer Schering establishes its own manufacturing capability.
Research and Development
We currently conduct research and development activities at our facilities in Bothell, Washington. We also engage in certain research, preclinical studies and clinical development efforts at third party laboratories and other institutions. Our primary research and development efforts are currently directed at the development and application of the proprietary TOCOSOL technology to TOCOSOL Paclitaxel and to a lesser extent, other various compounds where we can use our expertise or technology to improve either the safety or efficacy of oncology drugs.
Our research and development activities for the last three years can be divided into research, preclinical and clinical development programs primarily associated with TOCOSOL Paclitaxel as well as research and preclinical activities related to our other early stage pipeline development product candidates. The approximate costs associated with these programs for the last three fiscal years were as follows (in millions):
|
|
2006 |
|
2005 |
|
2004 |
|
||||
|
TOCOSOL Paclitaxel |
|
$ |
32.4 |
|
$ |
21.2 |
|
$ |
9.2 |
|
|
Other research & preclinical programs |
|
$ |
8.7 |
|
$ |
3.3 |
|
$ |
1.5 |
|
|
Total research & development |
|
$ |
41.1 |
|
$ |
24.5 |
|
$ |
10.7 |
|
We separately track all billable costs associated with TOCOSOL paclitaxel as it is our lead product candidate and has been partnered with Bayer Schering. Costs attributed to other research and preclinical projects largely represent our pipeline generating activities and are not tracked to the same level of precision. See Item 7. Managements Discussion and Analysis of Financial Condition and Results of Operations for further discussion of research and development spending trends.
Government Regulations Drug Approval Process
Regulation by governmental authorities in the U.S. and other countries is a significant factor in our ongoing research and development activities and in the production and marketing of our products. In order to undertake clinical tests, to produce and market products for human use, mandatory procedures and safety standards, established by the FDA in the U.S. and by comparable agencies in other countries, must be followed.
The standard process before a pharmaceutical agent may be marketed includes the following steps:
· Preclinical studies including laboratory evaluation and animal studies to test for initial safety and efficacy;
· Submission to national health authorities of an Investigational New Drug application (IND), or Clinical Trials Application (CTA) or equivalent dossier, which must be accepted by each national health authority before human clinical trials may commence in that country;
· Adequate and
well-controlled clinical trials to establish the safety and efficacy of the
drug in its intended population
and use(s);
· Submission to appropriate national and/or regional regulatory health authorities of a New Drug Application (NDA), or equivalent marketing authorization application, which application is not automatically accepted for review; and
· approval by appropriate regulatory health authorities of the marketing authorization application prior to any commercial sale or shipment of the drug in each country or jurisdiction.
7
In addition to obtaining regulatory health authority approvals for each product, each drug-manufacturing establishment for products to be sold in the U.S. must be registered by the FDA for each product that is manufactured at that facility. Manufacturing establishments are subject to inspections by the FDA and by other federal, state and local agencies and must comply with GMP requirements applicable to the production of pharmaceutical drug products. GMP requirements are enumerated in FDA regulations and guidance documents. The facilities, procedures, and operations of contract manufacturers must be determined to be adequate by the FDA before approval of commercial product manufacturing. Manufacturing facilities are subject to inspections by the FDA for compliance with GMP, licensing specifications, and other regulations. Failure to comply with regulations can result in fines, unanticipated compliance expenditures, recall or seizure of products, total or partial suspension of production and/or distribution, suspension of the FDAs review of NDAs, injunctions and criminal prosecution.
Preclinical studies include laboratory evaluation of the active drug substance and its formulation in animal studies to assess the potential safety and efficacy of the drug and its formulation. Prior to initiating the first clinical testing of a new drug product candidate, the results of the preclinical studies are submitted to regulatory health authorities as part of an IND or CTA, and must be accepted before the proposed clinical trial(s) can begin.
Clinical trials for cancer therapeutics involve the administration of the investigational drug product to patients with a defined disease state, under the supervision of a qualified principal investigator. Clinical trials are conducted in accordance with protocols that detail the objectives of the study, the parameters to be used to monitor safety and the efficacy criteria to be evaluated. Each protocol is submitted to regulatory health authorities as part of the IND/CTA, in each country where clinical trials using our investigational products are to be conducted. Each clinical study is approved and monitored by one or more independent Institutional Review Boards or Ethics Committees who consider, among other things, ethical factors, informed consent documents, the safety of human subjects and the possible liability of the institutions conducting a clinical study. The Institutional Review Board or Ethics Committee may require changes in the clinical trials protocol, which may delay initiation or completion of the study.
Clinical trials typically are conducted in three sequential phases, although the phases may overlap. In Phase 1, the initial introduction of the drug to humans, the drug is tested for acute safety and clinical pharmacology. Phase 2 trials involve more detailed evaluation of the safety and efficacy of the drug in patients with a defined disease or condition. Phase 3 trials consist of large scale evaluations of safety and efficacy of the investigational product compared to accepted standard therapy in a defined disease or condition.
The process of completing clinical testing and obtaining regulatory health authority approval for a new product takes a number of years and requires the expenditure of substantial resources. In the U.S., the FDA may grant full approval of a drug product for a particular indication or may grant approval conditioned on further post-marketing clinical trials. Regulatory health authorities may conclude that the data submitted in a marketing authorization application are not adequate to support an approval and may require further clinical and preclinical testing, re-submission of the marketing application, and further review. Even after initial approval has been obtained, further studies may be required to provide additional data about the approved indication, and further studies will be required to gain approval for the use of a product for clinical indications other than those for which the product was approved initially. Also, health authorities may require post-marketing testing and surveillance programs to monitor the drug products side effects.
Marketing of pharmaceutical products outside of the U.S. is subject to regulatory requirements that vary from country to country. In the European Union, the general trend has been towards coordination of common standards for clinical testing of new drug products. Centralized approval in the European Union is coordinated through the European Medicines Evaluation Agency, or EMEA.
The level of regulation outside of the U.S. and European Union varies widely. The time required to obtain regulatory approval from regulatory agencies in each country may be longer or shorter than that required for FDA or EMEA approval. In addition, in certain markets, reimbursement may be subject to governmentally mandated prices.
Many of the chemicals and compounds used in our research and development efforts are classified as hazardous materials under applicable federal, state and local environmental laws and regulations. We are subject to regulations under state and federal law regarding occupational safety, laboratory practices, handling and disposing of chemicals, environmental protection and hazardous substance control. We also will be subject to other possible future regulations of local, state, federal and other jurisdictions.
Competition
The healthcare industry in general is characterized by extensive research efforts, rapid technological change and intense competition. We believe that other pharmaceutical companies will compete with us in areas of research and development, acquisition of products and technology licenses, and the manufacturing and marketing of products that could potentially
8
compete with ours. We expect that competition will be based on safety, efficacy, ease of administration, breadth of approved indications, price, reimbursement and physician and patient acceptance.
Several other companies are developing enhanced delivery taxanes with a goal of delivering a more effective and tolerable therapy than Taxotere, Taxol and the approved generic paclitaxel-based products. On January 7, 2005, American Pharmaceutical Partners (now Abraxis BioScience) obtained FDA approval to market its paclitaxel-based product, ABRAXANE® (paclitaxel protein-bound particles for injectable suspension). In addition, Sanofi-aventis has a taxane product, Taxotere (docetaxel), which has a similar mechanism of action to paclitaxel and is marketed for the treatment of breast, non-small cell lung and prostate cancers. There are also a number of generic paclitaxel products, identical to Taxol, currently on the market.
We believe that our ability to successfully compete in the biotechnology and pharmaceutical industries will be based on our ability to do the following:
· Create and maintain advanced formulation technologies;
· Develop proprietary products;
· Attract and retain key scientific personnel;
· Obtain patent or other protection for products;
· Obtain required regulatory approvals; and
· Manufacture, market and or license our products alone or with collaborative partners.
Many of our competitors and potential competitors have substantially greater financial, technical and human resources than we do and have substantially greater experience in developing products, obtaining regulatory approvals and marketing and manufacturing products. Companies that complete clinical trials, obtain required regulatory approvals and commence commercial sales of their products before their competitors may achieve a significant competitive advantage if their products work through a similar mechanism as our products. In addition, other technologies or products may be developed that have an entirely different approach that would render our technology and products noncompetitive or obsolete.
Patents and Proprietary Rights
We consider the protection of our technology to be important to our business. In addition to seeking U.S. patent protection for our inventions, we are also seeking patent protection in other selected countries in order to broadly protect our proprietary rights. We also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position.
Our success will depend, in part, on our ability to obtain and defend patents and protect trade secrets. As of December 31, 2006, nine United States patents and five patents outside the U.S., one each in Canada, Taiwan, Mexico, Korea and India have been issued pertaining to our proprietary TOCOSOL technology. Additional patent applications are pending in the United States and counterpart filings have been made in Europe, Canada and key countries in Asia and Latin America.
The patent position of medical and pharmaceutical companies is highly uncertain and involves complex legal and factual questions. There can be no assurance that any claims which are included in pending or future patent applications will be issued, that any issued patents will provide us with competitive advantage or will not be challenged by third parties, or that existing or future patents of third parties will not have an adverse effect on our ability to commercialize our products. Furthermore, there can be no assurance that other companies will not independently develop similar products, duplicate any of our products or design around patents that may be issued to us. Litigation or administrative proceedings may be necessary to enforce any patents issued to us or to determine the scope and validity of others proprietary rights.
Our commercial success will depend in part on not infringing patents issued to competitors. There can be no assurance that patents belonging to competitors or others will not require us to alter our products or processes, pay licensing fees or cease development of our current or future products. Further, there can be no assurance that we will be able to license other technology that we may require at a reasonable cost or at all. Failure by us to obtain a license to any technology that we may require to commercialize our products could have a material adverse effect on our business, financial condition and results of operations.
We have obtained registration for our marks TOCOSOL® and Sonus Pharmaceuticals®, in the United States. There can be no assurance that the registered or unregistered trademarks or trade names of our company will not infringe upon third party rights or will be acceptable to regulatory agencies.
We also rely on unpatented trade secrets, proprietary know-how and continuing technological innovation, which we seek to protect, in part, by confidentiality agreements with our corporate partners, collaborators, employees and consultants in our
9
drug development research. There can be no assurance that these agreements will not be breached, that we will have adequate remedies for any breach, or that our trade secrets or know-how will not otherwise become known or be independently discovered by competitors. Further, there can be no assurance that we will be able to protect our trade secrets or that others will not independently develop substantially equivalent proprietary information and techniques.
Product Liability
The testing, marketing and sale of pharmaceutical products entails a risk of product liability claims by consumers and others. We currently maintain product liability insurance for our clinical trials with limits of $10 million per claim and in the aggregate, which we believe to be adequate for current non-commercial and Phase 3 applications of our products. In the event of a successful suit against us, the lack or insufficiency of insurance coverage could have a material adverse effect on our business and financial condition. Although we have never been subject to a product liability claim, there can be no assurance that the coverage limits of our insurance policies will be adequate or that one or more successful claims brought against us would not have a material adverse effect upon our business, financial condition and results of operations. If any of our products under development gain marketing approval from the FDA or other regulatory health authorities, there can be no assurance that adequate product liability insurance will be available, or if available, that it will be available at a reasonable cost. Any adverse outcome resulting from a product liability claim could have a material adverse effect on our business, financial condition and results of operations.
Employees
As of March 1, 2007, we had 61 employees, 44 engaged in research and development, regulatory, clinical and manufacturing activities, and 17 in business operations and administration. All of our employees are covered by confidentiality agreements. We consider our relations with our employees to be good, and none of our employees is a party to a collective bargaining agreement.
Sonus Pharmaceuticals was incorporated in California in October 1991 and subsequently reorganized as a Delaware corporation in September 1995. The Companys principal executive offices are located at 22026 20th Avenue SE, Bothell, Washington 98021, and its telephone number is (425) 487-9500. The Company makes its annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to these reports available on its website, at http://www.sonuspharma.com, free of charge as soon as practicable after filing with the SEC. All such reports are also available free of charge via EDGAR through the SEC website at www.sec.gov. In addition, the public may read and copy materials filed by the Company with the SEC at the SECs public reference room located at 450 Fifth St., N.W., Washington, D.C., 20549. Information regarding operation of the SECs public reference room can be obtained by calling the SEC at 1-800-SEC-0330.
ITEM 1A. RISK FACTORS
You should consider the risk below carefully in addition to other information contained in this report before engaging in any transaction involving shares of our common stock. If any of these risks occur, they could seriously harm our business, financial condition or results of operations. In such case, the trading price of our common stock could decline, and you may lose all or part of your investment.
Governmental regulatory requirements are lengthy and expensive and failure to obtain necessary approvals will prevent us or our partners from commercializing a product.
We are subject to uncertain governmental regulatory requirements and a lengthy approval process for our products prior to any commercial sales of our products. The development and commercial use of our products are regulated by the U.S. Food and Drug Administration, or FDA, the European Medicines Evaluation Agency, or EMEA, and comparable regulatory agencies in other countries. The regulatory approval process for new products is lengthy and expensive. Before we can submit an application to the FDA and comparable international agencies, the product candidate must undergo extensive testing, including animal studies and human clinical trials that can take many years and require substantial expenditures. Data obtained from such testing may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. In addition, changes in regulatory policy for product approval may cause additional costs in our efforts to secure necessary approvals.
Our product candidates are subject to significant uncertainty because they are in both early to late stages of development and are subject to regulatory approval. The results of preclinical and clinical testing of our products are uncertain and regulatory approval of our products may take longer or be more expensive than anticipated, which could have a material
10
adverse effect on our business, financial condition and results of operations. In June 2005, the FDA completed its review of the contents of the SPA for TOCOSOL Paclitaxel. The FDA has indicated to Sonus that NDA approval under 505(b)(2) will require either (i) demonstration of superior efficacy of TOCOSOL Paclitaxel as compared to Taxol; or (ii) demonstration of non-inferior efficacy of TOCOSOL Paclitaxel as compared to Taxol, and either a change of the approved label for Taxol and generic equivalents to include a weekly dosing schedule or availability of reviewable data from a Phase 3 trial demonstrating superior efficacy of Taxol using a weekly dosing schedule as compared to that of Taxol using the currently approved three-weekly dosing schedule.
We do not currently believe that the timing or cost of the Phase 3 trial or the NDA submission will be adversely affected by these requirements. The clinical trial Protocol and Statistical Analysis Plan approved under the SPA provide for sequential superiority analyses for efficacy of TOCOSOL Paclitaxel compared to Taxol, provided that we first demonstrate a non-inferior objective response rate; however, there can be no assurance that the Phase 3 clinical trial data will demonstrate that TOCOSOL Paclitaxel has efficacy that is non-inferior or superior to Taxol. Further, there can be no assurance that the approved label for Taxol or generics will be changed to provide for weekly dosing, although we do believe, based on several discussions with the FDA, that they are pursuing this change.
We have an agreement with the Cancer and Leukemia Group B Foundation (the CALGB) giving us the right to use data from the CALGB Study 9840, a Phase 3 trial comparing weekly dosing of Taxol to three-weekly dosing of Taxol in patients with metastatic breast cancer. In the event that TOCOSOL Paclitaxel does not achieve superior efficacy over Taxol in the Phase 3 trial or the approved label for Taxol is not changed to include a weekly dosing schedule, Bayer Schering and we plan to submit the CALGB 9840 data to the FDA as part of the TOCOSOL Paclitaxel NDA to support weekly dosing of Taxol as an appropriate reference arm in the ongoing pivotal Phase 3 trial. Based on the summary presentation of CALGB 9840 at the American Society of Clinical Oncology (ASCO) 2004 annual meeting, our analysis of the data set and discussions with the FDA, we believe that the data from this study should fulfill the FDAs requirement to submit a reviewable data set that compares weekly dosing of Taxol to three-weekly dosing of Taxol, and demonstrate the weekly regimen to be superior to the every three-weekly regimen. While there can be no assurance that the data obtained from this study will be sufficient to support the TOCOSOL Paclitaxel NDA, Sonus preliminary analysis of the CALGB 9840 data confirm the conclusions presented in the abstract at ASCO 2004. If the FDA does not accept the CALGB Study 9840 data as support for a weekly reference arm, substantial additional costs and time would be required before the NDA submission for TOCOSOL Paclitaxel.
In addition, there is pending litigation attacking the utilization of the 505(b)(2) regulatory strategy generally. There can be no assurance that such litigation will not be successful. A 505(b)(2) application permits us to rely upon the FDAs findings of safety and efficacy for a previously approved drug product without requiring us to obtain a right of reference from the original applicant. In addition to permitting reliance upon the FDAs prior findings of safety and effectiveness for previously approved drugs, section 505(b)(2) continues to allow reliance on third party data that is available in published literature and which establishes the safety and effectiveness of a drug. However, we are required to provide any additional clinical data necessary to demonstrate the safety and effectiveness of differences between the original drug and the 505(b)(2) drug, so while unnecessary duplication of preclinical and certain human studies is avoided, specific studies may be required to establish the relevance and applicability of prior findings for our particular product formulation. We cannot predict if or when any of our products under development will be commercialized.
We will need additional capital in the future to support the continued development and commercialization of TOCOSOL Paclitaxel, our obligations under the Collaboration and License Agreement with Bayer Schering and to fund continuing operations.
We expect that our cash requirements will continue to increase in future periods due to development costs associated with TOCOSOL Paclitaxel and other product candidates. We believe that existing cash, cash equivalents and marketable securities, in addition to future payments and cost sharing arrangements under our Collaboration and License Agreement with Bayer Schering, will be sufficient to fund operations through the second quarter of 2008. We will need additional capital in 2008 to complete the development and commercialization of TOCOSOL Paclitaxel, fund our obligations under the Collaborative License Agreement with Bayer Schering, fund the development of other product candidates and support our continuing operations. In addition to the supportive trials Sonus plans to conduct, it is anticipated that we will collaborate with Bayer Schering on additional studies. Under the terms of the Collaboration and License Agreement with Bayer Schering, we are also obligated to fund 50% of the costs of certain studies conducted by Bayer Schering for the U.S. The exact cost and timing of these studies is yet to be finalized. Our current estimate for the total cost of the Phase 3 trial is approximately $50 million. This estimate is our direct cost only and does not include any future billings from Bayer Schering. However, the scope, timing and costs of the Phase 3 clinical trial are difficult to determine with accuracy and these costs may vary significantly depending upon regulatory and other matters that are not within our control. Enrollment in the Phase 3 study was closed in November 2006 with 821 patients randomized. Should our clinical data support an NDA
11
submission based on the primary endpoint of objective response rate, we anticipate that the NDA, the contents of which are being developed in collaboration Bayer Schering, will be submitted by Bayer Schering in the second quarter of 2008. Our future capital requirements depend on many factors including:
· our ability to obtain and timing of payments under corporate partner agreements and/or debt or equity financings;
· our ability to obtain and timing of capital funding under equity or debt financing agreements;
· timing and costs of preclinical development, clinical trials and regulatory approvals;
· timing and amount of costs to support our obligations under the Collaboration and License Agreement with Bayer Schering;
· entering into new collaborative or product license agreements;
· timing and costs of technology transfer associated with manufacturing and supply agreements; and
· costs related to obtaining, defending and enforcing patents.
Any future debt or equity financing, if available, may result in substantial dilution to existing stockholders, and debt financing, if available, may include restrictive covenants.
If we fail to develop new products, then we may never realize revenue from product commercialization.
A key element of our business strategy is to utilize our technologies for the development and commercialization of products that utilize our proprietary TOCOSOL technology. Most of our attention and resources are directed to the development of our proprietary TOCOSOL technology, a technology that provides a novel approach to the formulation of water insoluble compounds for therapeutic applications. Significant expenditures in additional research and development, clinical testing, regulatory, manufacturing, and sales and marketing activities will be necessary in order for us to demonstrate the efficacy of our products, or commercialize any products developed with our technology. There can be no assurance that product candidates under development or any future products will be safe or efficacious. If the product candidates under development are ultimately ineffective in treating cancer, do not receive the necessary regulatory approvals or do not obtain commercial acceptance, we will incur additional losses, our accumulated deficit will increase and our business will be materially adversely affected.
Even if we are successful in developing our products, there is no assurance that such products will receive regulatory approval or that a commercially viable market will develop.
Our future prospects are heavily dependent on the results of the Phase 3 clinical testing of TOCOSOL Paclitaxel and subsequent commercialization should the product be approved by the FDA and other international regulatory agencies.
TOCOSOL Paclitaxel is the only product candidate we have in Phase 3 clinical testing. All other product candidates are in much earlier stages of development. Due to the significant costs and data performance uncertainties and other significant uncertainties involved in getting the other product candidates to Phase 3 clinical testing, our current prospects are substantially dependent on the outcome of the current Phase 3 trial with TOCOSOL Paclitaxel. There can also be no assurance that performance seen to-date for TOCOSOL Paclitaxel in Phase 1 and 2 clinical testing will be reflected in the final data for the current Phase 3 clinical trial, or that the product will be approved by the FDA or other international regulatory agencies, or achieve commercial success, if approved.
We have a history of operating losses which we expect will continue and we may never become profitable.
We have experienced significant accumulated losses since our inception, and are expected to incur net losses for the foreseeable future. These losses have resulted primarily from expenses associated with our research and development activities, including nonclinical and clinical trials, and general and administrative expenses. As of December 31, 2006, our accumulated deficit totaled $111.7 million. We anticipate that our operating losses will continue as we further invest in research and development for our products. We will not generate the majority of milestone or royalty revenues under our collaboration and license agreement with Bayer Schering unless and until we receive regulatory approvals, which are not likely to occur until 2009 and beyond. Even if we generate milestone and royalty revenues, there can be no assurance that we will be able to achieve or sustain profitability. Our results of operations have varied and will continue to vary significantly and depend on, among other factors:
· our ability to obtain and timing of payments under corporate partner agreements and/or debt or equity financings;
· timing and costs of preclinical development, clinical trials and regulatory approvals;
· timing and amount of costs to support our obligations under the Collaboration and License Agreement with Bayer Schering;
12
· drug discovery and research and development;
· entering into new collaborative or product license agreements for products in our pipeline;
· timing and costs of technology transfer associated with manufacturing and supply agreements; and
· costs related to obtaining, defending and enforcing patents.
We depend on third parties for funding, clinical development, manufacturing and distribution of TOCOSOL Paclitaxel.
We are dependent, and may in the future be dependent, on third parties for funding or performance of a variety of key activities including research, clinical development, manufacturing, marketing, sales and distribution of our products. Our current business strategy is to enter into agreements with third parties both to license rights to our potential products and to develop and commercialize new products. We executed an agreement with Bayer Schering for TOCOSOL Paclitaxel in October 2005. Under the Collaboration and License Agreement, Bayer Schering has a worldwide exclusive license to market and promote TOCOSOL Paclitaxel and is responsible for clinical development and regulatory activities outside of the U.S. If these arrangements with Bayer Schering or other third parties are terminated or the collaborations are not successful, we will be required to identify alternative sources of funding to finance research, clinical development, manufacturing, marketing, sales and/or distribution. Our inability to secure additional funding would have a material adverse effect on our business, financial condition and results of operations. Our success depends in part upon the performance by these collaborators of their responsibilities under these arrangements. We have no control over the resources that our partners may devote to the development and commercialization of products under these collaborations and our partners may fail to conduct their collaborative activities successfully or in a timely manner. On April 13, 2006, a wholly owned subsidiary of Bayer AG, a German corporation, or Bayer, submitted a formal tender offer to the stockholders of Schering AG to purchase all of the outstanding shares of Schering AG. The acquisition was essentially complete as of December 31, 2006 with only some minor activities remaining. We are not aware of any material effect the acquisition has had on our business, financial condition or results of operations but there can be no assurance that the acquisition will not have a material effect in the future.
If we lose our key personnel or are unable to attract and retain qualified scientific and management personnel, we may be unable to become profitable.
We are highly dependent on our key executives, including Michael A. Martino, President & Chief Executive Officer and Alan Fuhrman, Senior Vice President & Chief Financial Officer. We do not have employment agreements in place with these key executives nor do we maintain any key person life insurance coverage on these persons. The loss of any of these key executives or the inability to recruit and retain qualified scientific personnel to perform research and development and qualified management personnel could have a material adverse effect on our business, financial condition and results of operations. There can be no assurance that we will be able to attract and retain such personnel on acceptable terms, if at all, given the competition for experienced scientists and other personnel among numerous medical and pharmaceutical companies, universities and research institutions.
Future U.S. or international legislative or administrative actions also could prevent or delay regulatory approval of our products.
Even if regulatory approvals are obtained, they may include significant limitations on the indicated uses for which a product may be marketed. A marketed product also is subject to continual FDA, EMEA and other regulatory agency review and regulation. Later discovery of previously unknown problems or failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market, as well as possible civil or criminal sanctions. In addition, if marketing approval is obtained, the FDA, EMEA or other regulatory agency may require post-marketing testing and surveillance programs to monitor the products efficacy and side effects. Results of these post-marketing programs may prevent or limit the further marketing of a product.
The development of pharmaceutical products in general and the development of paclitaxel reformulations in particular is extremely competitive, and if we fail to compete effectively, it would negatively impact our business.
Competition in the development of pharmaceutical products is intense and expected to increase. We also believe that other medical and pharmaceutical companies will compete with us in the areas of research and development, acquisition of products and technology licenses, and the manufacturing and marketing of our products. Success of products in these fields will be based primarily on:
· efficacy;
· safety;
· price;
13
· ease of administration;
· breadth of approved indications; and
· physician, healthcare payor and patient acceptance.
Several other companies are developing paclitaxel reformulations with a goal of delivering a more effective and tolerable therapy than the approved paclitaxel products. Some of these products are further in development than TOCOSOL Paclitaxel and may achieve regulatory approval before our product. On January 7, 2005, Abraxis BioScience (formerly American Pharmaceutical Partners) obtained FDA approval to market its paclitaxel-based product, ABRAXANE (paclitaxel protein-bound particles for injectable suspension). In addition, sanofi-aventis has a taxane product, Taxotere (docetaxel), which is similar to paclitaxel and is marketed for the treatment of breast, non-small cell lung and prostate cancers. There are also a number of generic paclitaxel products, identical to Taxol, currently on the market.
Many of our competitors and potential competitors, including large pharmaceutical, chemical and biotechnology concerns and universities and other research institutions, have substantially greater financial, technical and human resources than we do and have substantially greater experience in developing products, obtaining regulatory approvals and marketing and manufacturing medical products. Accordingly, these competitors may succeed in obtaining FDA approval for their products more rapidly than we do. In addition, other technologies or products may be developed that have an entirely different approach that would render our technology and products noncompetitive or obsolete. If we fail to compete effectively, it would have a material adverse effect on our business, financial condition and results of operations.
We rely on third party suppliers and manufacturers to produce products that we develop and failure to retain such suppliers and manufacturers would adversely impact our ability to commercialize our products.
We currently rely on third parties to supply the chemical ingredients necessary for our drug product candidates. We have entered into supply agreements for the supply of GMP grade paclitaxel, which is the active pharmaceutical ingredient in TOCOSOL Paclitaxel. The chemical ingredients for our products are manufactured by a limited number of vendors. The inability of these vendors to supply medical-grade materials to us could delay the manufacturing of, or cause us to cease the manufacturing of our products. We also rely on third parties to manufacture our products for research and development and clinical trials. TEVA Pharmaceuticals USA (TEVA) is our primary manufacturer of TOCOSOL Paclitaxel for clinical studies. On March 2, 2006, in accordance with the Collaboration and License Agreement with Bayer Schering, Bayer Schering exercised their right to assume responsibility for the manufacturing of TOCOSOL Paclitaxel. In June 2006, we entered into a clinical supply agreement with Bayer Schering to provide clinical supplies of TOCOSOL Paclitaxel to Bayer Schering until such time as Bayer Schering establishes its own manufacturing capability. Suppliers and manufacturers of our products must operate under GMP regulations, as required by the FDA, and there are a limited number of contract manufacturers that operate under GMP regulations. GMP are enumerated in FDA regulations and guidance documents. The facilities, procedures, and operations of our contract manufacturers must be determined to be adequate by the FDA before approval of product manufacturing. Manufacturing facilities are subject to inspections by the FDA for compliance with GMP, licensing specifications, and other FDA regulations. Failure to comply with FDA and other governmental regulations can result in fines, unanticipated compliance expenditures, recall or seizure of products, total or partial suspension of production and/or distribution, suspension of the FDAs review of NDAs, injunctions and criminal prosecution. Any of these actions could have a material adverse effect on us. Our reliance on independent manufacturers involves a number of other risks, including the absence of adequate capacity, the unavailability of, or interruptions in, access to necessary manufacturing processes and reduced control over delivery schedules. If our manufacturers are unable or unwilling to continue manufacturing our products in required volumes or have problems with commercial scale-up, we will have to identify acceptable alternative manufacturers. The use of a new manufacturer may cause significant interruptions in supply if the new manufacturer has difficulty manufacturing products to our specifications. Further, the introduction of a new manufacturer may increase the variation in the quality of our products.
If we fail to secure adequate intellectual property protection or become involved in an intellectual property dispute, it could significantly harm our financial results and ability to compete.
Our success will depend, in part, on our ability to obtain and defend patents and protect trade secrets. As of December 31, 2006, we held nine United States patents and five patents outside the U.S., one each in Canada, Korea, Taiwan, Mexico and India pertaining to our proprietary TOCOSOL technology. We hold one additional United States patent directed to other technologies. Additional patent applications are pending in the United States and counterpart filings have been made in Europe, Canada and key countries in Asia and Latin America. The patent position of medical and pharmaceutical companies is highly uncertain and involves complex legal and factual questions. There can be no assurance that any claims which are included in pending or future patent applications will be issued, that any issued patents will provide us with competitive advantages or will not be challenged by third parties, or that the existing or future patents of third parties will not have an
14
adverse effect on our ability to commercialize our products. Furthermore, there can be no assurance that other companies will not independently develop similar products, duplicate any of our products or design around patents that may be issued to us. Litigation may be necessary to enforce any patents issued to us or to determine the scope and validity of others proprietary rights in court or administrative proceedings. Any litigation or administrative proceeding could result in substantial costs to us and distraction of our management. An adverse ruling in any litigation or administrative proceeding could have a material adverse effect on our business, financial condition and results of operations.
Our commercial success will depend in part on not infringing patents issued to competitors.
There can be no assurance that patents belonging to competitors will not require us to alter our products or processes, pay licensing fees or cease development of our current or future products. Any litigation regarding infringement could result in substantial costs to us and distraction of our management, and any adverse ruling in any litigation could have a material adverse effect on our business, financial condition and results of operations. Further, there can be no assurance that we will be able to license other technology that we may require at a reasonable cost or at all. Failure by us to obtain a license to any technology that we may require to commercialize our products could result in the termination of the Collaboration and License Agreement with Bayer Schering and would have a material adverse effect on our business, financial condition and results of operations. In addition, to determine the priority of inventions and the ultimate ownership of patents, we may participate in interference, reissue or re-examination proceedings conducted by the U.S. Patent and Trademark Office or in proceedings before international agencies with respect to any of our existing patents or patent applications or any future patents or applications, any of which could result in loss of ownership of existing, issued patents, substantial costs to us and distraction of our management.
Reimbursement procedures and future healthcare reform measures are uncertain and may adversely impact our ability to successfully sell pharmaceutical products.
Our ability to successfully sell any pharmaceutical products will depend in part on the extent to which government health administration authorities, private health insurers and other organizations will reimburse patients for the costs of future pharmaceutical products and related treatments. In the United States, government and other third-party payors have sought to contain healthcare costs by limiting both coverage and the level of reimbursement for new pharmaceutical products approved for marketing by the FDA. In some cases, these payors may refuse to provide any coverage for uses of approved products to treat medical conditions even though the FDA has granted marketing approval. Healthcare reform may increase these cost containment efforts. We believe that managed care organizations may seek to restrict the use of new products, delay authorization to use new products or limit coverage and the level of reimbursement for new products. Internationally, where national healthcare systems are prevalent, little if any funding may be available for new products, and cost containment and cost reduction efforts can be more pronounced than in the United States.
If our products are not accepted by the medical community our business will suffer.
Commercial sales of our proposed products will substantially depend upon the products efficacy and on their acceptance by the medical community. Widespread acceptance of our products will require educating the medical community as to the benefits and reliability of the products. Our proposed products may not be accepted, and, even if accepted, we are unable to estimate the length of time it would take to gain such acceptance.
The businesses in which we engage have a risk of product liability, and in the event of a successful suit against us, our business could be severely harmed.
The testing, marketing and sale of pharmaceutical products entails a risk of product liability claims by consumers and others. We currently maintain product liability insurance for our clinical trials with limits of $10 million per claim and in the aggregate, which we believe to be adequate for current non-commercial and Phase 3 applications of our products. In the event of a successful suit against us, the lack or insufficiency of insurance coverage could have a material adverse effect on our business and financial condition.
Since we use hazardous materials in our business, we may be subject to claims relating to improper handling, storage or disposal of these materials.
Our research and development activities involve the controlled use of hazardous materials, chemicals and various radioactive compounds. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by state and federal regulations, the risk of
15
accidental contamination or injury from these materials cannot be eliminated completely. In the event of such an accident, we could be held liable for any damages that result and any such liability not covered by insurance could exceed our resources. Compliance with environmental laws and regulations may be expensive, and current or future environmental regulations may impair our research, development or production efforts.
Market volatility may affect our stock price and the value of an investment in our common stock may be subject to sudden decreases.
The trading price for our common stock has been, and we expect it to continue to be, volatile. The price at which our common stock trades depends upon a number of factors, including our historical and anticipated operating results, preclinical and clinical trial results, market perception of the prospects for biotechnology companies as an industry sector and general market and economic conditions, some of which are beyond our control. Factors such as fluctuations in our financial and operating results, changes in government regulations affecting product approvals, reimbursement or other aspects of our or our competitors businesses, FDA review of our product development activities, the results of preclinical studies and clinical trials, announcements of technological innovations or new commercial products by us or our competitors, developments concerning key personnel and our intellectual property rights, significant collaborations or strategic alliances and publicity regarding actual or potential performance of products under development by us or our competitors could also cause the market price of our common stock to fluctuate substantially. In addition, the stock market has from time to time experienced extreme price and volume fluctuations. These broad market fluctuations may lower the market price of our common stock. Moreover, during periods of stock market price volatility, share prices of many biotechnology companies have often fluctuated in a manner not necessarily related to the companies operating performance. Also, biotechnology or pharmaceutical stocks may be volatile even during periods of relative market stability. Accordingly, our common stock may be subject to greater price volatility than the stock market as a whole.
Failure to satisfy NASDAQ Global Market listing requirements may result in our common stock being delisted from The NASDAQ Global Market.
Our common stock is currently listed on The NASDAQ Global Market under the symbol SNUS. For continued inclusion on The NASDAQ Global Market, we must maintain among other requirements stockholders equity of at least $10.0 million, a minimum bid price of $1.00 per share and a market value of our public float of at least $5.0 million; or market capitalization of at least $50 million, a minimum bid price of $1.00 per share and a market value of our public float of at least $15.0 million. As of December 31, 2006, we had stockholders equity of approximately $43.0 million. In the event that we fail to satisfy the listing standards on a continuous basis, our common stock may be removed from listing on The NASDAQ Global Market. If our common stock were delisted from The NASDAQ Global Market, our common stock may be transferred to the NASDAQ Capital Market if we satisfy the listing criteria for the NASDAQ Capital Market or trading of our common stock, if any, may be conducted in the over-the-counter market in the so-called pink sheets or, if available, the National Association of Securities Dealers Electronic Bulletin Board. Consequently, broker-dealers may be less willing or able to sell and/or make a market in our common stock. Additionally, an investor would find it more difficult to dispose of, or to obtain accurate quotations for the price of, our common stock. As a result of a delisting, it may become more difficult for us to raise funds through the sale of our securities.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
We currently lease approximately 27,000 square feet of laboratory and office space in a single facility near Seattle, Washington. The lease expires in July 2007 and includes an option to extend the term of the lease for three years. We did not exercise the lease option and instead signed a new lease agreement for a larger facility also near Seattle, Washington. The new lease involves approximately 37,000 square feet of laboratory and office space in a single facility. The lease has a 10 year term and includes two options to renew for additional 5 year periods. We plan to move into the new facility in the fourth quarter of 2007.
16
From time to time, the Company may be involved in litigation relating to claims arising out of our operations in the normal course of business. The Company currently is not a party to any legal proceedings, the adverse outcome of which, in managements opinion, individually or in the aggregate, would have a material adverse effect on the Companys results of operations or financial position.
ITEM 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
No matters were submitted to a vote of security holders during the fourth quarter of the year ended December 31, 2006.
ITEM 5. MARKET FOR THE REGISTRANTS COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock first began trading on the Nasdaq National Market under the symbol SNUS on October 12, 1995. No cash dividends have been paid on the common stock, and we do not anticipate paying any cash dividends in the foreseeable future. As of March 1, 2007, there were approximately 160 stockholders of record and approximately 6,400 beneficial stockholders of our Common Stock. The high and low sales prices of our common stock as reported by Nasdaq Global Market (formerly the NASDAQ National Market) for the periods indicated are as follows:
|
|
High |
|
Low |
|
|||
|
2005 |
|
|
|
|
|
||
|
First Quarter |
|
$ |
4.50 |
|
$ |
2.56 |
|
|
Second Quarter |
|
3.85 |
|
2.39 |
|
||
|
Third Quarter |
|
5.04 |
|
3.33 |
|
||
|
Fourth Quarter |
|
5.28 |
|
3.77 |
|
||
|
|
|
|
|
|
|
||
|
2006 |
|
|
|
|
|
||
|
First Quarter |
|
$ |
6.92 |
|
$ |
4.85 |
|
|
Second Quarter |
|
6.28 |
|
4.40 |
|
||
|
Third Quarter |
|
5.15 |
|
4.25 |
|
||
|
Fourth Quarter |
|
6.32 |
|
4.51 |
|
||
The information required by this item regarding equity compensation plan information is set forth in Part III, Item 12 of this Annual Report filed on Form 10-K. We made no purchases of equity securities during the fourth quarter of the year ended December 31, 2006.
17
Stock Performance Graph
Set forth below is a line graph comparing the cumulative stockholder return on the Companys Common Stock with the cumulative total return of the Nasdaq Composite Index and the Nasdaq Pharmaceutical Index for the five year period that commenced December 31, 2001 and ended on December 31, 2006.
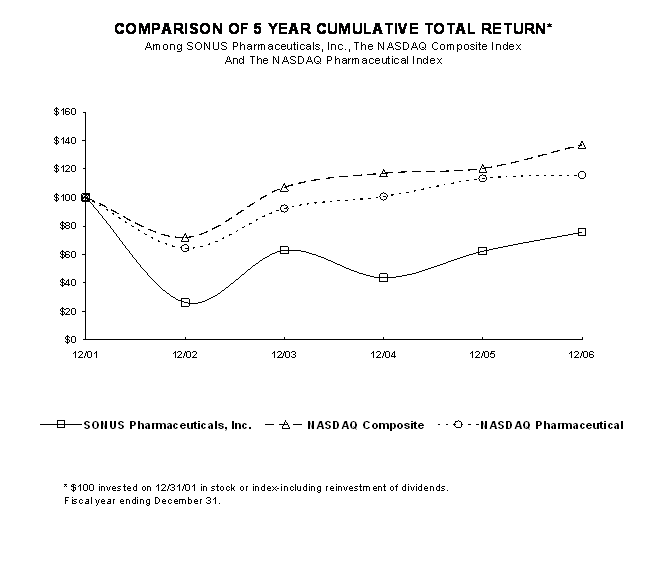
18
ITEM 6. SELECTED FINANCIAL DATA
The data set forth below should be read in conjunction with Item 7, Managements Discussion and Analysis of Financial Condition and Results of Operations and the Financial Statements and Notes thereto appearing at Item 8 of this report.
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2006 |
|
2005 |
|
2004 |
|
2003 |
|
2002 |
|
|||||
|
|
|
(in thousands, except per share data) |
|
|||||||||||||
|
Statements of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Total revenue |
|
$ |
22,392 |
|
$ |
8,254 |
|
$ |
|
|
$ |
25 |
|
$ |
25 |
|
|
Operating expenses |
|
$ |
48,679 |
|
$ |
30,064 |
|
$ |
16,576 |
|
$ |
10,663 |
|
$ |
12,199 |
|
|
Net loss |
|
$ |
(23,551 |
) |
$ |
(21,097 |
) |
$ |
(16,311 |
) |
$ |
(10,467 |
) |
$ |
(11,636 |
) |
|
Net loss per share: |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Basic |
|
$ |
(0.68 |
) |
$ |
(0.88 |
) |
$ |
(0.81 |
) |
$ |
(0.68 |
) |
$ |
(0.86 |
) |
|
Diluted |
|
$ |
(0.68 |
) |
$ |
(0.88 |
) |
$ |
(0.81 |
) |
$ |
(0.68 |
) |
$ |
(0.86 |
) |
|
Shares used in calculation of net loss per share |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Basic |
|
34,730 |
|
24,027 |
|
20,169 |
|
15,504 |
|
13,564 |
|
|||||
|
Diluted |
|
34,730 |
|
24,027 |
|
20,169 |
|
15,504 |
|
13,564 |
|
|||||
|
|
|
December 31, |
|
|||||||||||||
|
|
|
2006 |
|
2005 |
|
2004 |
|
2003 |
|
2002 |
|
|||||
|
|
|
(in thousands) |
|
|||||||||||||
|
Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Cash, cash equivalents and marketable securities |
|
$ |
58,278 |
|
$ |
49,318 |
|
$ |
20,580 |
|
$ |
19,664 |
|
$ |
16,334 |
|
|
Accounts receivable from Bayer Schering Pharma AG |
|
$ |
8,044 |
|
$ |
7,057 |
|
$ |
|
|
$ |
|
|
$ |
|
|
|
Total assets |
|
$ |
68,493 |
|
$ |
57,914 |
|
$ |
22,571 |
|
$ |
21,468 |
|
$ |
17,934 |
|
|
Current liabilities |
|
$ |
19,910 |
|
$ |
11,242 |
|
$ |
3,255 |
|
$ |
1,794 |
|
$ |
1,938 |
|
|
Long-term liabilities |
|
$ |
5,541 |
|
$ |
11,408 |
|
$ |
239 |
|
$ |
364 |
|
$ |
272 |
|
|
Stockholders equity |
|
$ |
43,042 |
|
$ |
35,264 |
|
$ |
19,077 |
|
$ |
19,310 |
|
$ |
15,724 |
|
ITEM 7. MANAGEMENTS DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This report contains certain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, and we intend that such forward-looking statements be subject to the safe harbors created thereby. Examples of these forward-looking statements include, but are not limited to:
· timing and amount of future contractual payments, product revenue and operating expenses;
· progress and preliminary results of clinical trials;
· our anticipated future capital requirements and the terms of any capital financing agreements;
· anticipated regulatory filings, requirements and future clinical trials; and
· market acceptance of our products and the estimated potential size of these markets.
While these forward-looking statements made by us are based on our current beliefs and judgments, they are subject to risks and uncertainties that could cause actual results to vary from the projections in the forward-looking statements. You should consider the risks below carefully in addition to other information contained in this report before engaging in any transaction involving shares of our common stock. If any of these risks occur, they could seriously harm our business, financial condition or results of operations. In such case, the trading price of our common stock could decline, and you may lose all or part of your investment.
19
The discussion and analysis set forth in this document contains trend analysis, discussions of regulatory status and other forward-looking statements. Actual results could differ materially from those projected in the forward-looking statement as a result of the following factors, among others:
· uncertainty of governmental regulatory requirements and lengthy approval process;
· future capital requirements and uncertainty of payments under corporate partnerships or additional funding through either debt or equity financings;
· dependence on the development and commercialization of products;
· future prospects heavily dependent on results of the Phase 3 trial for TOCOSOL Paclitaxel and subsequent commercialization should the product be approved by the FDA;
· history of operating losses and uncertainty of future financial results;
· dependence on third parties for funding, clinical development, manufacturing and distribution;
· dependence on key employees;
· uncertainty of U.S. or international legislative or administrative actions;
· competition and risk of competitive new products;
· limited manufacturing experience and dependence on a limited number of contract manufacturers and suppliers;
· ability to obtain and defend patents, protect trade secrets and avoid infringing patents held by third parties;
· limitations on third-party reimbursement for medical and pharmaceutical products;
· acceptance of our products by the medical community;
· potential for product liability issues and related litigation;
· potential for claims arising from the use of hazardous materials in our business;
· volatility in the value of our common stock; and
· continued listing on the NASDAQ Global Market (formerly NASDAQ National Market).
MD&A Overview
In Managements Discussion and Analysis of Financial Condition and Results of Operations we explain the general financial condition and the results of operations for our Company, including:
· An overview of our business;
· Results of operations and why those results are different from the prior year; and
· The capital resources we currently have and possible sources of additional funding for future capital requirements.
Overview
Sonus Pharmaceuticals is focused on the development of cancer drugs that are designed to provide better efficacy, safety and tolerability, and are easier to use. Our business strategy is as follows:
· Develop proprietary formulations of cancer drugs and new chemical entities utilizing our TOCOSOL® technology; and
· Identify and acquire products/technologies that are complementary to our focus in oncology in order to broaden our business and market opportunities.
Results of Operations
As of December 31, 2006, our accumulated deficit was approximately $111.7 million. We expect to incur substantial additional operating losses over the next several years. Such losses have been and will continue to principally be the result of various costs associated with our discovery and research and development programs. Substantially all of our working capital in recent years has resulted from equity financings and payments under corporate partnership agreements. Our ability to achieve a consistent, profitable level of operations depends in large part on obtaining regulatory approval for TOCOSOL Paclitaxel as well as future product candidates in addition to successfully manufacturing and marketing those products once they are approved. Even if we are successful in the aforementioned activities our operations may not be profitable. In addition, payments under corporate partnerships and licensing arrangements are subject to significant fluctuations in both timing and amount. Therefore, our operating results for any period may fluctuate significantly and may not be comparable to the operating results for any other period.
Collaboration and License Agreement with Bayer Schering
On October 17, 2005, we entered into a Collaboration and License Agreement with Bayer Schering, a German corporation, pursuant to which, among other things, we granted Bayer Schering an exclusive, worldwide license to
20
TOCOSOL Paclitaxel. Bayer Schering paid us an upfront license fee of $20 million and pays us for research and development services performed equal to 50% of eligible research and development costs related to TOCOSOL Paclitaxel (in certain cases the reimbursement rate is 100%). In addition, Bayer Schering may pay us (i) product milestone payments of up to $132 million upon the achievement of certain U.S., European Union and Japanese clinical and regulatory milestones, (ii) sales milestone payments of up to $35 million upon the achievement of certain annual worldwide net sales, and (iii) upon commercialization, royalties ranging between 15-30% of annual net sales in the U.S., with the exact percentage to be determined based on the achievement of certain annual net sales thresholds, and royalties equal to 15% of the annual net sales outside the U.S. The parties have agreed to a U.S. development program consisting of the ongoing Phase 3 pivotal trial for FDA NDA approval in metastatic breast cancer, and trials to support launch of TOCOSOL Paclitaxel and planned trials for additional indications. We have retained an option to exercise co-promotion rights in the U.S. and have also granted Bayer Schering the right of first negotiation on the novel camptothecin molecule Sonus is currently developing that is in Phase 1 clinical testing.
On March 2, 2006, in accordance with the Collaboration and License Agreement with Bayer Schering, Bayer Schering exercised their right to assume responsibility for the manufacturing of TOCOSOL Paclitaxel. In June 2006, we entered into a clinical supply agreement with Bayer Schering to provide clinical supplies of TOCOSOL Paclitaxel to Bayer Schering until such time as Bayer Schering establishes its own manufacturing capability.
On April 13, 2006, a wholly owned subsidiary of Bayer AG, a German corporation, or Bayer, submitted a formal tender offer to the stockholders of Schering AG to purchase all of the outstanding shares of Schering AG. The acquisition was essentially complete as of December 31, 2006 with only some minor activities remaining. We are not aware of any material effect the acquisition has had on our business, financial condition or results of operations but there can be no assurance that the acquisition will not have a material effect in the future.
Years Ended December 31, 2006 and December 31, 2005
Our revenue was $22.4 million for the year ended December 31, 2006 as compared with $8.3 million for 2005. Revenue in 2006 and 2005 was fully attributable to the collaboration agreement with Bayer Schering. We recognized $5.5 million in amortization of the upfront license fee and an additional $16.9 million in research and development reimbursements under the terms of our agreement with Bayer Schering. Amortization of the upfront fee will continue until the end of the development period for TOCOSOL Paclitaxel which is currently estimated at the end of 2008 or the currently estimated date for FDA approval assuming no further research is required and the results of the Phase 3 trial successfully meet its endpoints. Research and development funding will also continue during this time. This estimate is subject to change as facts and circumstances surrounding our Phase 3 trial for TOCOSOL Paclitaxel. We expect revenue to decline substantially in 2007 as reimbursements from Bayer Schering associated with the Phase 3 trial begin to wind down and Bayer Schering takes over full manufacturing of TOCOSOL Paclitaxel. These reimbursements provide the majority of our revenue.
Our research and development (R&D) expenses were $41.1 million for the year ended December 31, 2006 compared with $24.5 million for 2005. The 2006 increase was primarily the result of the spending associated with the Phase 3 clinical trial for TOCOSOL Paclitaxel including both clinical and drug supply and manufacturing costs (both control and study drug) as well as costs associated with the implementation of SFAS 123R. We expect R&D expenses to increase slightly in 2007 primarily on increased spending for preclinical activity associated with new product development as well as TOCOSOL Paclitaxel, offset in part by lower Phase 3 trial costs for TOCOSOL Paclitaxel as that trial begins to trend down.
Our general and administrative (G&A) expenses were $7.6 million for the year ended December 31, 2006 compared with $5.6 million for 2005. The 2006 increase was primarily attributed to costs associated with the implementation of SFAS 123R as well as market research conducted on TOCOSOL Paclitaxel as that product moves closer to FDA submission. We believe that G&A expenses will increase in 2007 on increased administrative support for TOCOSOL Paclitaxel and other product candidates as we continue to grow the business.
Our total operating expenses in 2007 are expected to increase from 2006 levels due to expected increased preclinical activity for both TOCOSOL Paclitaxel and new product development and higher G&A expenses. We estimate that R&D spending will comprise approximately 80%-85% of the anticipated spending in 2007. A significant portion of the R&D spending will be devoted to the Phase 3 and other supportive clinical trials for TOCOSOL Paclitaxel. These estimates and actual expenses are subject to change depending on many factors, including unforeseen expansion of study size or duration, complications in conducting or completing studies when the study begins, changes in FDA requirements, increased material costs and other factors.
Our interest income, net of interest expense, was $2.8 million for the year ended December 31, 2006 compared with $708,000 for 2005. The 2006 increase was due primarily to higher levels of invested cash in 2006 in addition to generally higher interest rates throughout 2006.
21
The Company had no income tax expense in 2006, 2005 or 2004 as it had incurred pretax losses.
Years Ended December 31, 2005 and December 31, 2004
Our revenue was $8.3 million for the year ended December 31, 2005 as compared with $0 million for 2004. Revenue in 2005 was fully attributable to the collaboration agreement with Bayer Schering.
Our research and development (R&D) expenses were $24.5 million for the year ended December 31, 2005 compared with $10.7 million for 2004. The 2005 increase was primarily the result of the spending associated with the Phase 3 clinical trial for TOCOSOL Paclitaxel including both clinical and drug supply and manufacturing costs (both control and study drug).
Our general and administrative (G&A) expenses were $5.6 million for the year ended December 31, 2005 compared with $5.9 million for 2004. The 2005 decrease was primarily attributed to approximately $1.0 million in costs related to the termination of our acquisition of Synt:em as well as increased personnel, business development and Sarbanes-Oxley compliance costs in 2004.
Our interest income, net of interest expense, was $708,000 for the year ended December 31, 2005 compared with $265,000 for 2004. The 2005 increase was due primarily to higher levels of invested cash in 2005 in addition to generally higher interest rates throughout 2005.
The Company had no income tax expense in 2005 or 2004 as it had incurred significant losses and has significant net operating loss carryforwards.
Liquidity and Capital Resources
We have historically financed operations with proceeds from equity financings and payments under collaboration agreements with third parties. At December 31, 2006, we had cash, cash equivalents and marketable securities totaling $58.3 million compared to $49.3 million at December 31, 2005. The increase was primarily due to the net proceeds of $28.6 million from the issuance of common stock on May 2, 2006 and payments of $15.7 million received from Bayer Schering under the collaboration agreement of which $7.2 million related to 2005 receivables. These increases were offset by the net loss for 2006 of $23.6 million, payment of 2005 incentive bonuses in 2006 of $1.2 million and other timing differences.
Net cash used in operating activities for the years ended December 31, 2006, 2005 and 2004, was $19.7 million, $8.4 million and $14.6 million, respectively. Expenditures in all periods were primarily a result of R&D expenses, including clinical trial costs, and G&A expenses in support of our operations and product development activities primarily related to TOCOSOL Paclitaxel and to a lesser extent other potential product candidates. We believe that G&A expenses will increase in 2007 on increased administrative support for TOCOSOL Paclitaxel and other product candidates as we continue to grow the business. We expect R&D expenses to increase slightly in 2007 primarily on increased spending for preclinical activity associated with new product development as well as TOCOSOL Paclitaxel, offset in part by lower Phase 3 trial costs for TOCOSOL Paclitaxel as that trial begins to trend down. We recorded $22.4 million in revenue in 2006, $8.3 million in 2005 and none in 2004. We expect revenue to decline substantially in 2007 as reimbursements from Bayer Schering associated with the Phase 3 trial begin to wind down and Bayer Schering takes over full manufacturing of TOCOSOL Paclitaxel. We paid no corporate income taxes in any of the periods presented.
Net cash provided by (used in) investing activities for the years ended December 31, 2006, 2005 and 2004, was ($23.0) million, $20.1 million and ($2.6) million, respectively. The cash used in investing activities in 2006 was primarily related to purchases of marketable securities as we began moving a portion of our portfolio to marketable securities due to better interest rate opportunities and our cash position in general. The cash provided by investing activities in 2005 was primarily related to sales and maturities of marketable securities occurring in the normal course of business. The cash used in investing activities in 2004 was primarily related to purchases of marketable securities offset in part by sales and maturities of marketable securities. In all of the years presented, we raised money in the capital markets and/or through corporate partnerships which was invested in either cash equivalent or marketable securities.
Net cash provided by financing activities for the years ended December 31, 2006, 2005 and 2004, was $29.1 million, $37.2 million and $16.0 million, respectively. The net cash provided by financing activities in 2006 and 2005 primarily related to proceeds from equity financing, payments received from Bayer Schering under our Collaboration and License Agreement, the exercise of common stock warrants and the issuance of common stock under employee benefit plans. The net cash provided by financing activities in 2004 primarily related to proceeds from equity financings, exercise of common stock warrants and the issuance of common stock under employee benefit plans.
We expect that our cash requirements will continue to increase in future periods due to the development and commercialization costs associated with TOCOSOL Paclitaxel and other product candidates. We believe that existing cash,
22
cash equivalents and marketable securities, in addition to payments pending and cost sharing arrangements under our Collaboration and License Agreement with Bayer Schering, will be sufficient to fund operations through the second quarter of 2008. In addition to the supportive trials Sonus plans to conduct, it is anticipated that we will collaborate with Bayer Schering on additional studies. Under the terms of the Collaboration and License Agreement with Bayer Schering, we are also obligated to fund 50% of the costs of certain studies conducted by Bayer Schering for the U.S. The exact cost and timing of these studies is yet to be finalized. In addition, the scope, timing and costs of the Phase 3 clinical trial are difficult to determine with accuracy and these costs may vary significantly depending upon regulatory and other matters that are not within our control. Our current estimate for the total cost of the Phase 3 clinical trial is approximately $50 million. This estimate is our direct cost only and does not include any future billings from Bayer Schering. Enrollment in the Phase 3 study concluded in November. We will need additional capital in 2008 to support the continued development and commercialization of TOCOSOL Paclitaxel, our obligations under the Collaboration and License Agreement with Bayer Schering, the development of other product candidates and to fund continuing operations. Should our clinical data support an NDA submission based on the primary endpoint of objective response rate, we anticipate that the NDA, the contents of which are being developed in collaboration with Bayer Schering, will be submitted by Bayer Schering in the second quarter of 2008. Our future capital requirements depend on many factors including:
· our ability to obtain and timing of payments under corporate partner agreements and/or debt or equity financings;
· timing and costs of preclinical development, clinical trials and regulatory approvals;
· timing and amount of costs to support our obligations under the Collaboration and License Agreement with Bayer Schering;
· timing and cost of drug discovery and research and development;
· entering into new collaborative or product license agreements for products in our pipeline;
· timing and costs of technology transfer associated with manufacturing and supply agreements; and
· costs related to obtaining, defending and enforcing patents.
We have contractual obligations in the form of operating leases and leasehold financing arrangements, which expire in 2007. The Company signed a new facility lease in November 2006. The new facility lease has a term of 10 years with a provision for two additional five year renewals. The estimated commencement date for the new lease is October 2007. The following table summarizes our contractual obligations under these agreements, including interest as of December 31, 2006:
|
Contractual Obligations |
|
Total |
|
Less than |
|
1-3 years |
|
3-5 years |
|
More than 5 |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
Lease financing obligations |
|
$ |
15,197 |
|
$ |
15,197 |
|
$ |
|
|
$ |
|
|
$ |
|
|
|
Operating lease obligations |
|
20,325,529 |
|
1,096,483 |
|
3,569,760 |
|
3,752,720 |
|
11,906,566 |
|
|||||
|
Total |
|
$ |
20,340,726 |
|
$ |
1,111,680 |
|
$ |
3,569,760 |
|
$ |
3,752,720 |
|
$ |
11,906,566 |
|
Under the Collaboration and License Agreement with Bayer Schering, we are obligated to fund 50% of the costs of certain studies conducted by Bayer Schering. As these additional studies have not yet been finalized, no dollar amounts have been disclosed above.
Critical Accounting Policies and Estimates
We prepare our financial statements in conformity with accounting principles generally accepted in the United States of America. As such, we are required to make certain estimates, judgments and assumptions that we believe are reasonable based upon the information available. These estimates and assumptions affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the periods presented. Actual results could differ significantly from those estimates under different assumptions and conditions. We believe that the following discussion addresses our most critical accounting estimates which are those that are most important to the portrayal of our financial condition and results of operations and which require our most difficult and subjective judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain. We also have other policies that we consider key accounting policies; however, these policies do not meet the definition of critical accounting estimates, because they do not generally require us to make estimates or judgments which are difficult or subjective.
· Cash, Cash Equivalents and Marketable Securities. We consider investments in highly liquid instruments purchased with a remaining maturity at purchase of 90 days or less to be cash equivalents. Investments with a remaining maturity at purchase in excess of 90 days are classified as marketable securities. The amounts are recorded at cost, which approximate fair market value. Our cash equivalents and marketable securities consist principally of commercial paper, money market securities, repurchase agreements, corporate bonds/notes and government agency securities. We have classified our entire investment portfolio as available-for-sale. Available-for-sale securities are
23
carried at fair value, with unrealized gains and losses reported as a separate component of stockholders equity and included in accumulated other comprehensive income. The amortized cost of investments is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization and accretion are included in interest income. Interest earned on securities is included in interest income. We consider marketable securities with maturity greater than twelve months long-term and maturity less than twelve months short-term.
· Revenue Recognition. Since inception, we have generated revenue from collaborative agreements, licensing fees and from the assignment of developed and patented technology. These arrangements may include upfront non-refundable payments, development milestone payments, payments for research and development services performed and product sales royalties or revenue. Our revenue recognition policies are based on the requirements of SEC Staff Accounting Bulletin No. 104 Revenue Recognition, and, for contracts with multiple deliverables, we allocate arrangement consideration based on the fair value of the elements under guidance from Emerging Issues Task Force Issue 00-21 (EITF 00-21), Revenue Arrangements with Multiple Deliverables. Under EITF 00-21, revenue arrangements with multiple deliverables are divided into separate units of accounting and revenue is allocated to these units based upon relative fair values with revenue recognition criteria considered separately for each unit.
Nonrefundable upfront technology license fees, for product candidates where we are providing continuing services related to product development, are deferred and recognized as revenue over the estimated development period.
The timing and amount of revenue that we recognize from licenses of technology, either from upfront fees or milestones where we are providing continuing services related to product development, is primarily dependent upon our estimates of the development period. We define the development period as the point from which research activities commence up to FDA approval of our submission assuming no further research is necessary. As product candidates move through the development process, it is necessary to revise these estimates to consider changes to the product development cycle, such as changes in the clinical development plan, regulatory requirements, or various other factors, many of which may be outside of our control. Should our clinical development plans change, as a result of regulatory or other matters, we would adjust our development period estimates accordingly. The impact on revenue of changes in our estimates and the timing thereof is recognized prospectively over the remaining estimated product development period. Revenue from research and development services performed under collaboration agreements is generally recognized in the period when the services are performed. Payments received in excess of amounts earned are recorded as deferred revenue.
· Research and Development Expenses. Pursuant to SFAS No. 2 Accounting for Research and Development Costs, our research and development costs are expensed as incurred. Research and development expenses include, but are not limited to, payroll and personnel expenses, lab expenses, clinical trial and related clinical manufacturing costs, facilities and overhead costs. Clinical trial expenses, which are included in research and development expenses and represent a significant portion of our research and development expenditures, represent obligations resulting from our contracts with various clinical research organizations in connection with conducting clinical trials for our product candidates. We recognize expenses for these contracted activities based on a variety of factors, including actual and estimated labor hours, clinical site initiation activities, patient enrollment rates, estimates of external costs and other activity-based factors. We believe that this method best approximates the efforts expended on a clinical trial with the expenses we record. We adjust clinical expense estimates when actual results are available.
· Stock-based Compensation. We adopted the requirements of SFAS 123R, Share-Based Payment, effective January 1, 2006, utilizing the modified prospective method. We use the Black-Scholes-Merton option pricing model as the most appropriate fair-value method for our awards and recognize compensation cost on a straight-line basis over our awards vesting periods in accordance with the provisions of SFAS 123R. In valuing our options using the Black-Scholes-Merton option pricing model, we make assumptions about risk-free interest rates, dividend yields, volatility and weighted average expected lives of the options. Risk-free interest rates are derived from United States treasury securities as of the option grant date. Dividend yields are based on our historical dividend payments, which have been zero to date. Volatility is derived from the historical volatility of our common stock as traded on NASDAQ. Forfeiture rates are estimated using historical actuals over the estimated life of the option grant. These rates are adjusted on a quarterly basis and any change in compensation expense is recognized in the period of the change. The weighted average expected lives of the options is based on historical experience of option exercises and the average vesting option schedule. For unvested awards granted prior to the adoption date, compensation expense is based on the original grant date fair value measurement under SFAS 123. We currently believe that the assumptions used to generate those fair values are appropriate.
24
Recent Accounting Pronouncements
In June 2006, the Financial Accounting Standards Board (FASB) issued FIN 48, Accounting for Uncertainty in Income Taxes, an interpretation of FASB Statement 109. FIN 48 prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. The Interpretation is effective for fiscal years beginning after December 15, 2006. We are required to adopt this interpretation in the first quarter of 2007. The company continues to evaluate the impact of FIN 48 on its financial position and results of operations. At this time, the effects of adoption have not yet been determined.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest rate risk:
The market risk inherent in our marketable securities portfolio represents the potential loss arising from adverse changes in interest rates. If market rates hypothetically increase immediately and uniformly by 100 basis points from levels at December 31, 2006, the decline in the fair value of the investment portfolio would not be material. Given the short-term nature of our investment portfolio, we do not expect our operating results or cash flows to be affected to any significant degree by a sudden change in market interest rates.
Foreign currency exchange risk:
We are exposed to risks associated with foreign currency transactions on certain contracts denominated in foreign currencies (primarily Euro denominated contracts) and we have not hedged these amounts. As our unhedged foreign currency transactions fluctuate, our earnings might be negatively affected. Accordingly, changes in the value of the U.S. dollar relative to the Euro might have an adverse effect on our reported results of operations and financial condition, and fluctuations in exchange rates might harm our reported results and accounts from period to period. The impact of foreign currency fluctuations related to realized gains and losses during the past three years has not been material.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
INDEX TO FINANCIAL STATEMENTS:
25
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Sonus Pharmaceuticals, Inc.
We have audited the accompanying balance sheets of Sonus Pharmaceuticals, Inc. as of December 31, 2006 and 2005, and the related statements of operations, stockholders equity, and cash flows for each of the three years in the period ended December 31, 2006. These financial statements are the responsibility of the Companys management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Sonus Pharmaceuticals, Inc. at December 31, 2006 and 2005, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2006, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the effectiveness of Sonus Pharmaceuticals, Inc.s internal control over financial reporting as of December 31, 2006, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 13, 2007 expressed an unqualified opinion thereon.
As discussed in Note 1 to the financial statements, in 2006, Sonus Pharmaceuticals, Inc. changed its method of accounting for share-based payments in accordance with the guidance provided in Statement of Financial Accounting Standards No. 123(R), Share-Based Payments.
|
|
ERNST & YOUNG LLP |
|
Seattle, Washington |
|
|
March 13, 2007 |
|
26
Sonus
Pharmaceuticals, Inc.
Balance Sheets
|
|
|
December 31, |
|
||||
|
|
|
2006 |
|
2005 |
|
||
|
ASSETS |
|
|
|
|
|
||
|
Current assets: |
|
|
|
|
|
||
|
Cash and cash equivalents |
|
$ |
35,771,784 |
|
$ |
49,317,845 |
|
|
Marketable securities |
|
22,506,086 |
|
|
|
||
|
Accounts receivable from Bayer Schering Pharma AG |
|
8,043,771 |
|
7,056,640 |
|
||
|
Other current assets |
|
524,470 |
|
341,787 |
|
||
|
Total current assets |
|
66,846,111 |
|
56,716,272 |
|
||
|
|
|
|
|
|
|
||
|
Equipment, furniture and leasehold improvements, net |
|
1,186,174 |
|
1,006,403 |
|
||
|
Long term receivable from Bayer Schering Pharma AG |
|
|
|
87,500 |
|
||
|
Other assets |
|
460,717 |
|
103,739 |
|
||
|
|
|
|
|
|
|
||
|
Total assets |
|
$ |
68,493,002 |
|
$ |
57,913,914 |
|
|
|
|
|
|
|
|
||
|
LIABILITIES AND STOCKHOLDERS EQUITY |
|
|
|
|
|
||
|
Current liabilities: |
|
|
|
|
|
||
|
Accounts payable |
|
$ |
898,486 |
|
$ |
1,260,513 |
|
|
Accounts payable to Bayer Schering Pharma AG |
|
1,473,050 |
|
|
|
||
|
Accrued expenses |
|
11,928,124 |
|
4,407,844 |
|
||
|
Deferred revenue from Bayer Schering Pharma AG |
|
5,545,919 |
|
5,545,920 |
|
||
|
Current portion of lease obligations |
|
14,763 |
|
27,410 |
|
||
|
Other current liabilities |
|
50,029 |
|
|
|
||
|
Total current liabilities |
|
19,910,371 |
|
11,241,687 |
|
||
|
|
|
|
|
|
|
||
|
Deferred revenue from Bayer Schering Pharma AG, less current portion |
|
5,540,694 |
|
11,086,612 |
|
||
|
Lease obligations, less current portion |
|
|
|
14,763 |
|
||
|
Other liabilities |
|
|
|
307,060 |
|
||
|
|
|
|
|
|
|
||
|
Commitments and contingencies |
|
|
|
|
|
||
|
Stockholders equity: |
|
|
|
|
|
||
|
Preferred stock, $.001 par value: 5,000,000 shares authorized; no shares outstanding |
|
|
|
|
|
||
|
Common stock, $.001 par value: 75,000,000 shares authorized; 36,853,974 and 30,565,746 shares issued and outstanding in 2006 and 2005, respectively |
|
154,780,939 |
|
123,443,666 |
|
||
|
Accumulated deficit |
|
(111,738,669 |
) |
(88,187,373 |
) |
||
|
Accumulated other comprehensive income (loss) |
|
(333 |
) |
7,499 |
|
||
|
Total stockholders equity |
|
43,041,937 |
|
35,263,792 |
|
||
|
|
|
|
|
|
|
||
|
Total liabilities and stockholders equity |
|
$ |
68,493,002 |
|
$ |
57,913,914 |
|
See accompanying notes.
27
Sonus
Pharmaceuticals, Inc.
Statements of Operations
|
|
|
Year Ended December 31, |
|
|||||||
|
|
|
2006 |
|
2005 |
|
2004 |
|
|||
|
Revenue: |
|
|
|
|
|
|
|
|||
|
Collaboration revenue from Bayer Schering Pharma AG |
|
$ |
22,391,858 |
|
$ |
8,254,483 |
|
$ |
|
|
|
|
|
|
|
|
|
|
|
|||
|
Operating expenses: |
|
|
|
|
|
|
|
|||
|
Research and development |
|
41,102,276 |
|
24,493,651 |
|
10,706,223 |
|
|||
|
General and administrative |
|
7,576,276 |
|
5,570,051 |
|
5,869,331 |
|
|||
|
Total operating expenses |
|
48,678,552 |
|
30,063,702 |
|
16,575,554 |
|
|||
|
|
|
|
|
|
|
|
|
|||
|
Operating loss |
|
(26,286,694 |
) |
(21,809,219 |
) |
(16,575,554 |
) |
|||
|
|
|
|
|
|
|
|
|
|||
|
Other income (expense): |
|
|
|
|
|
|
|
|||
|
Other (expense) income |
|
(112,490 |
) |
4,160 |
|
|
|
|||
|
Interest income |
|
2,850,872 |
|
714,866 |
|
289,587 |
|
|||
|
Interest expense |
|
(2,984 |
) |
(6,824 |
) |
(24,625 |
) |
|||
|
Total other income, net |
|
2,735,398 |
|
712,202 |
|
264,962 |
|
|||
|
|
|
|
|
|
|
|
|
|||
|
Net loss |
|
$ |
(23,551,296 |
) |
$ |
(21,097,017 |
) |
$ |
(16,310,592 |
) |
|
|
|
|
|
|
|
|
|
|||
|
Basic and diluted net loss per share |
|
$ |
(0.68 |
) |
$ |
(0.88 |
) |
$ |
(0.81 |
) |
|
|
|
|
|
|
|
|
|
|||
|
Shares used in calculation of basic and diluted net loss per share |
|
34,729,930 |
|
24,027,127 |
|
20,169,258 |
|
|||
See accompanying notes.
28
Sonus
Pharmaceuticals, Inc.
Statements of Stockholders Equity
|
|
|
Common Stock |
|
Accumulated |
|
Accumulated |
|
|
|
||||||
|
|
|
Shares |
|
Amount |
|
Deficit |
|
Income (Loss) |
|
Total |
|
||||
|
Balance at January 1, 2004 |
|
17,957,452 |
|
$ |
70,085,299 |
|
$ |
(50,779,764 |
) |
$ |
4,148 |
|
$ |
19,309,683 |
|
|
Comprehensive income (loss): |
|
|
|
|
|
|
|
|
|
|
|
||||
|
Net loss |
|
|
|
|
|
(16,310,592 |
) |
|
|
(16,310,592 |
) |
||||
|
Change in unrealized gain (loss) on investments |
|
|
|
|
|
|
|
(38,791 |
) |
(38,791 |
) |
||||
|
Comprehensive loss |
|
|
|
|
|
|
|
|
|
(16,349,383 |
) |
||||
|
Issuance of common stock under employee benefit plans |
|
150,628 |
|
259,093 |
|
|
|
|
|
259,093 |
|
||||
|
Exercise of common stock warrants |
|
344,715 |
|
1,409,884 |
|
|
|
|
|
1,409,884 |
|
||||
|
Issuance of common stock (net of offering costs of $777,096) |
|
2,900,000 |
|
14,447,904 |
|
|
|
|
|
14,447,904 |
|
||||
|
Balance at December 31, 2004 |
|
21,352,795 |
|
86,202,180 |
|
(67,090,356 |
) |
(34,643 |
) |
19,077,181 |
|
||||
|
Comprehensive income (loss): |
|
|
|
|
|
|
|
|
|
|
|
||||
|
Net loss |
|
|
|
|
|
(21,097,017 |
) |
|
|
(21,097,017 |
) |
||||
|
Change in unrealized gain (loss) on investments |
|
|
|
|
|
|
|
42,142 |
|
42,142 |
|
||||
|
Comprehensive loss |
|
|
|
|
|
|
|
|
|
(21,054,875 |
) |
||||
|
Issuance of common stock under employee benefit plans |
|
86,082 |
|
185,117 |
|
|
|
|
|
185,117 |
|
||||
|
Exercise of common stock warrants |
|
575,000 |
|
2,351,750 |
|
|
|
|
|
2,351,750 |
|
||||
|
Issuance of common stock and common stock warrants (net of offering costs of $1,180,669) |
|
8,551,869 |
|
34,704,619 |
|
|
|
|
|
34,704,619 |
|
||||
|
Balance at December 31, 2005 |
|
30,565,746 |
|
123,443,666 |
|
(88,187,373 |
) |
7,499 |
|
35,263,792 |
|
||||
|
Comprehensive income (loss): |
|
|
|
|
|
|
|
|
|
|
|
||||
|
Net loss |
|
|
|
|
|
(23,551,296 |
) |
|
|
(23,551,296 |
) |
||||
|
Change in unrealized gain (loss) on investments |
|
|
|
|
|
|
|
(7,832 |
) |
(7,832 |
) |
||||
|
Comprehensive loss |
|
|
|
|
|
|
|
|
|
(23,559,128 |
) |
||||
|
Issuance of common stock under employee benefit plans |
|
84,553 |
|
292,113 |
|
|
|
|
|
292,113 |
|
||||
|
Stock-based compensation expense |
|
|
|
2,173,379 |
|
|
|
|
|
2,173,379 |
|
||||
|
Exercise of common stock warrants |
|
73,675 |
|
301,330 |
|
|
|
|
|
301,330 |
|
||||
|
Issuance of common stock (net of offering costs of $2,079,549) |
|
6,130,000 |
|
28,570,451 |
|
|
|
|
|
28,570,451 |
|
||||
|
Balance at December 31, 2006 |
|
36,853,974 |
|
$ |
154,780,939 |
|
$ |
(111,738,669 |
) |
$ |
(333 |
) |
$ |
43,041,937 |
|
See accompanying notes.
29
Sonus
Pharmaceuticals, Inc.
Statements of Cash Flows
|
|
|
Year Ended December 31, |
|
|||||||
|
|
|
2006 |
|
2005 |
|
2004 |
|
|||
|
Operating activities: |
|
|
|
|
|
|
|
|||
|
Net loss |
|
$ |
(23,551,296 |
) |
$ |
(21,097,017 |
) |
$ |
(16,310,592 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|||
|
Depreciation |
|
609,615 |
|
592,825 |
|
552,277 |
|
|||
|
Non-cash stock-based compensation |
|
2,173,379 |
|
|
|
|
|
|||
|
Accretion of net discount on securities |
|
(301,312 |
) |
(6,669 |
) |
(36,798 |
) |
|||
|
Gain on sale of capital equipment |
|
|
|
(4,160 |
) |
|
|
|||
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|||
|
Accounts receivable from Bayer Schering Pharma AG |
|
(987,131 |
) |
(7,056,640 |
) |
|
|
|||
|
Other current assets |
|
(182,683 |
) |
117,039 |
|
(311,742 |
) |
|||
|
Long term receivable from Bayer Schering Pharma AG |
|
87,500 |
|
|
|
|
|
|||
|
Other long term assets |
|
(356,978 |
) |
(139,739 |
) |
|
|
|||
|
Accounts payable |
|
(362,027 |
) |
445,310 |
|
618,325 |
|
|||
|
Accounts payable to Bayer Schering Pharma AG |
|
1,473,050 |
|
|
|
|
|
|||
|
Accrued expense |
|
7,520,280 |
|
2,046,338 |
|
915,437 |
|
|||
|
Deferred revenue from Bayer Schering Pharma AG |
|
(5,545,919 |
) |
16,632,532 |
|
|
|
|||
|
Other current liabilities |
|
50,029 |
|
|
|
|
|
|||
|
Other liabilities |
|
(307,060 |
) |
110,968 |
|
(47,532 |
) |
|||
|
Net cash used in operating activities |
|
(19,680,553 |
) |
(8,359,213 |
) |
(14,620,625 |
) |
|||
|
|
|
|
|
|
|
|
|
|||
|
Investing activities: |
|
|
|
|
|
|
|
|||
|
Purchases of capital equipment and leasehold improvements |
|
(789,386 |
) |
(119,443 |
) |
(426,001 |
) |
|||
|
Proceeds from sale of capital equipment |
|
|
|
4,160 |
|
|
|
|||
|
Purchases of marketable securities |
|
(22,585,008 |
) |
|
|
(31,830,775 |
) |
|||
|
Proceeds from sales of marketable securities |
|
|
|
7,360,968 |
|
8,198,719 |
|
|||
|
Proceeds from maturities of marketable securities |
|
372,402 |
|
12,851,484 |
|
21,421,000 |
|
|||
|
Net cash (used in) provided by investing activities |
|
(23,001,992 |
) |
20,097,169 |
|
(2,637,057 |
) |
|||
|
|
|
|
|
|
|
|
|
|||
|
Financing activities: |
|
|
|
|
|
|
|
|||
|
Proceeds from issuance of common stock and common stock warrants under equity financings, net of issuance costs |
|
28,570,451 |
|
34,704,619 |
|
14,447,904 |
|
|||
|
Proceeds from exercise of common stock warrants |
|
301,330 |
|
2,351,750 |
|
1,409,884 |
|
|||
|
Proceeds from issuance of common stock under employee benefit plans |
|
292,113 |
|
185,117 |
|
259,093 |
|
|||
|
Payments on lease obligations |
|
(27,410 |
) |
(78,444 |
) |
(151,369 |
) |
|||
|
Net cash provided by financing activities |
|
29,136,484 |
|
37,163,042 |
|
15,965,512 |
|
|||
|
|
|
|
|
|
|
|
|
|||
|
Change in cash and cash equivalents for the year |
|
(13,546,061 |
) |
48,900,998 |
|
(1,292,170 |
) |
|||
|
Cash and cash equivalents at beginning of year |
|
49,317,845 |
|
416,847 |
|
1,709,017 |
|
|||
|
|
|
|
|
|
|
|
|
|||
|
Cash and cash equivalents at end of year |
|
$ |
35,771,784 |
|
$ |
49,317,845 |
|
$ |
416,847 |
|
|
|
|
|
|
|
|
|
|
|||
|
Supplemental cash flow information: |
|
|
|
|
|
|
|
|||
|
Interest paid |
|
$ |
2,984 |
|
$ |
6,824 |
|
$ |
24,625 |
|
See accompanying notes.
30
Sonus Pharmaceuticals, Inc.
1. Description of Business and Summary of Accounting Policies
Overview
Sonus Pharmaceuticals is focused on the development of oncology drugs that provide better therapeutic alternatives for cancer patients, including improved efficacy, safety, tolerability and are more convenient to use. Our business strategy is as follows:
· develop proprietary formulations of cancer drugs utilizing our TOCOSOL® technology;
· develop novel formulations of oncology related drugs; and
· identify and acquire products/technologies that are complementary to our focus in oncology in order to broaden our business and market opportunities.
Liquidity
The Company has historically experienced recurring losses from operations which have generated an accumulated deficit of $111.7 million through December 31, 2006. For the year ended December 31, 2006, the Company used $19.7 million of cash to fund operations. At December 31, 2006, the Company had cash, cash equivalents and marketable securities of $58.3 million, and working capital of $46.9 million.
The Company expects that its cash requirements will continue to increase in future periods due to the projected development costs associated with TOCOSOL Paclitaxel and other product candidates. However, the Company believes that existing cash, cash equivalents and marketable securities, in addition to future payments and cost sharing arrangements under our Collaboration and License Agreement with Bayer Schering, will be sufficient to fund operations through the second quarter of 2008.
Cash and Cash Equivalents
Cash and cash equivalents consist of highly liquid investments with a maturity of three months or less at the date of purchase. Included as a component of cash equivalents are $35.6 million and $23.9 million of securities purchased under overnight repurchase agreements as of December 31, 2006 and 2005, respectively.
Fair Value of Financial Instruments
The carrying amounts of certain of the Companys financial instruments, principally cash and cash equivalents, and marketable securities approximate fair value due to their short maturities.
Marketable Securities
The Company classifies the marketable securities portfolio as available-for-sale, and such securities are stated at fair value based on quoted market prices, with the unrealized gains and losses included as a component of accumulated other comprehensive loss. Interest earned on securities available-for-sale is included in interest income. The carrying value of debt securities in this category is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization and accretion are included in interest income. Realized gains and losses and declines in value judged to be other than temporary on securities available-for-sale also are included in interest income. The cost of securities sold is based on the specific identification method.
Concentrations of Credit Risk
The Company invests its excess cash in accordance with investment guidelines, which limit the credit exposure to any one financial institution other than securities issued by the U.S. government. The guidelines also specify that the financial instruments are issued by institutions with strong credit ratings. These securities generally mature within one year or less and in some cases are not collateralized. The company is subject to credit risk on its receivables from its collaborative partner.
Revenue Recognition
Since inception, we have generated revenue from collaborative agreements, licensing fees and from the assignment of developed and patented technology. These arrangements may include upfront non-refundable payments, development milestone payments, payments for research and development services performed and product sales royalties or revenue. Our
31
revenue recognition policies are based on the requirements of SEC Staff Accounting Bulletin No. 104 Revenue Recognition, and, for contracts with multiple deliverables, we allocate arrangement consideration based on the fair value of the elements under guidance from Emerging Issues Task Force Issue 00-21 (EITF 00-21), Revenue Arrangements with Multiple Deliverables. Under EITF 00-21, revenue arrangements with multiple deliverables are divided into separate units of accounting and revenue is allocated to these units based upon relative fair values with revenue recognition criteria considered separately for each unit.
Nonrefundable upfront technology license fees, for product candidates where we are providing continuing services related to product development, are deferred and recognized as revenue over the estimated development period.
The timing and amount of revenue that we recognize from licenses of technology, either from upfront fees or milestones where we are providing continuing services related to product development, is primarily dependent upon our estimates of the development period. We define the development period as the point from which research activities commence up to FDA approval of our submission assuming no further research is necessary. As product candidates move through the development process, it is necessary to revise these estimates to consider changes to the product development cycle, such as changes in the clinical development plan, regulatory requirements, or various other factors, many of which may be outside of our control. Should our clinical development plans change, as a result of regulatory or other matters, we would adjust our development period estimates accordingly. The impact on revenue of changes in our estimates and the timing thereof is recognized prospectively over the remaining estimated product development period. Revenue from research and development services performed under collaboration agreements is generally recognized in the period when the services are performed. Payments received in excess of amounts earned are recorded as deferred revenue.
Research and Development Costs
Research and development (R&D) costs including personnel costs, supplies, depreciation and other indirect costs are expensed as incurred. Costs are expensed the earlier of when amounts are due or when services are performed. R&D expenses consist of independent R&D costs and costs associated with collaborative R&D arrangements.
Equipment, Furniture and Leasehold Improvements
Equipment, furniture and leasehold improvements are stated at cost. Depreciation of equipment is provided using the straight-line basis generally over three years for equipment and 5 years for furniture and fixtures which represents the estimated useful life of the assets. Leasehold improvements are amortized over the lesser of the economic useful lives of the improvements or the term of the related lease. The current lease has less than one year remaining. Repair and maintenance costs are expensed as incurred.
Segment Information
The Company follows the requirements of SFAS No. 131, Disclosure About Segments of an Enterprise and Related Information. The Company has one operating segment, the development of oncology drugs.
Stock-Based Compensation
The Company adopted the requirements of SFAS No. 123 (revised 2004), Share-Based Payment, (or SFAS 123R) on January 1, 2006, utilizing the modified prospective method. The Company uses the Black-Scholes-Merton option pricing model as the most appropriate fair-value method for its awards and recognizes compensation cost on a straight-line basis over its awards vesting periods in accordance with the provisions of SFAS 123R. In valuing its options using the Black-Scholes-Merton option pricing model, the Company makes assumptions about risk-free interest rates, dividend yields, volatility and weighted average expected lives, including estimated forfeiture rates, of the options. Risk-free interest rates are derived from United States treasury securities as of the option grant date. Dividend yields are based on the Companys historical dividend payments, which have been zero to date. Volatility is derived from the historical volatility of the Companys common stock as traded on NASDAQ. Forfeiture rates are estimated using historical actual forfeiture rates that resulted over the estimated life of the option grant for options granted as of the beginning of the forfeiture measurement period. These rates are adjusted on a quarterly basis and any change in compensation expense is recognized in the period of the change. The weighted average expected life of the options is based on historical experience of option exercises and the average vesting option schedule. In November 2005, the FASB Staff Position No. 123R-3, Transition Election Related to Accounting for the Tax Effects of Share-Based Payment Awards. The Company has adopted the simplified method to calculate the beginning balance of the additional paid-in-capital (or APIC) pool of excess tax benefit, and to determine the subsequent effect on the APIC pool and the Consolidated Statements of Cash Flows of the tax effects of stock-based compensation awards that were outstanding upon our adoption of FAS 123R.
For unvested awards granted prior to the adoption date, compensation expense is based on the original grant date fair value measurement under SFAS 123, Accounting for Stock-Based Compensation.
32
Income Taxes
The Company utilizes the liability method of accounting for income taxes as required by SFAS No. 109, Accounting for Income Taxes. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and tax reporting bases of assets and liabilities and are measured using enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. Valuation allowances are established when necessary to reduce deferred tax assets to the amounts expected to be realized. Due to uncertainty of the Companys ability to generate taxable income, a full valuation allowance has been established as of December 31, 2006.
Comprehensive Income
In accordance with Statement of Financial Accounting Standard No. 130, Reporting Comprehensive Income (SFAS 130), the Company has reported comprehensive income, defined as net income (loss) plus other comprehensive income, in the Statements of Stockholders Equity. The total of other accumulated comprehensive income consists of unrealized gains and losses on certain cash equivalent and marketable securities.
Per Share Data
Basic net loss per share is based on the weighted average number of common shares outstanding. Diluted net loss per share is based on the weighted average number of common shares and dilutive potential common shares. Dilutive potential common shares are calculated under the treasury stock method and consist of unexercised stock options and warrants.
Use of Estimates and Reclassifications
The preparation of financial statement in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
Certain reclassifications of prior period amounts have been made to our consolidated financial statements to conform to the current period presentation.
Recent Accounting Pronouncements
In June 2006, the Financial Accounting Standards Board (FASB) issued FIN 48, Accounting for Uncertainty in Income Taxes, an interpretation of FASB Statement 109. FIN 48 prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. The Interpretation is effective for fiscal years beginning after December 15, 2006. We are required to adopt this interpretation in the first quarter of 2007. The Company continues to evaluate the impact of FIN 48 on its financial position and results of operations. At this time, the effects of adoption have not yet been determined.
2. Collaboration and License Agreement with Bayer Schering
On October 17, 2005, the Company entered into a Collaboration and License Agreement with Bayer Schering, a German corporation, pursuant to which, among other things, the Company granted Bayer Schering an exclusive, worldwide license to its TOCOSOL Paclitaxel anti-cancer product candidate (the Product). With respect to the Product, Bayer Schering paid Sonus an upfront license fee of $20 million and pays Sonus for research and development services performed equal to 50% of eligible Product research and development costs (in certain cases the reimbursement rate is 100%). In addition, Bayer Schering may pay Sonus (i) product milestone payments of up to $132 million upon the achievement of certain U.S., European Union and Japanese clinical and regulatory milestones, (ii) sales milestone payments of up to $35 million upon the achievement of certain annual worldwide net sales, and (iii) upon commercialization, royalties ranging between 15-30% of annual net sales in the U.S., with the exact percentage to be determined based on the achievement of certain annual net sales thresholds, and royalties equal to 15% of the annual net sales outside the U.S. The parties have agreed to a development program consisting of the ongoing Phase 3 pivotal trial in metastatic breast cancer, and trials to support launch of the Product and planned trials for additional indications. The Company has retained an option to exercise co-promotion rights in the U.S. and has also granted Bayer Schering the right of first negotiation on the novel camptothecin molecule Sonus is currently developing that is in Phase 1 clinical testing. In connection with the Collaboration and Licensing Agreement, the Company and an affiliate of Bayer Schering entered into a Securities Purchase Agreement whereby the Company sold 3,900,000 shares of common stock for an aggregate of $15.7 million and warrants to purchase 975,000 shares of common stock for an aggregate purchase price of $122,000.
33
During the year ended December 31, 2006, the Company recognized revenue of $5.5 million as amortization of the upfront license fee and an additional $16.9 million related to research and development services performed for the Phase 3 trial for TOCOSOL Paclitaxel and related drug supply and manufacturing costs. The Company expects to recognize revenue related to amortization of the upfront fee and cost reimbursements through the end of the development period which is currently estimated as the end of 2008. The estimated development period represents the currently estimated date for FDA approval assuming no further research is required and the results of the Phase 3 trial successfully meet its endpoints. As the full clinical development program for TOCOSOL Paclitaxel, including other potential indications and geographic locations is still being finalized in collaboration with Bayer Schering, we cannot estimate the total costs or expected reimbursements at this time. The Company reduced the revenue to be recognized over the development period related to the $20 million upfront license payment by $2.3 million. This represented the excess fair value of the warrants purchased by an affiliate of Bayer Schering above the amount paid in connection with its equity investment in Sonus. This adjustment was made because both the equity investment and the upfront payment were considered to be a single unit of accounting. As of December 31, 2006, the Company had $11.1 million in deferred revenue related to the unamortized upfront payment (net of the adjustment for the warrant valuation) as well as $8.0 million in receivables from Bayer Schering on its balance sheet.
On March 2, 2006, in accordance with the Collaboration and License Agreement with Bayer Schering, Bayer Schering exercised their right to assume responsibility for the manufacturing of TOCOSOL Paclitaxel. In June 2006, we entered into a clinical supply agreement with Bayer Schering to provide clinical supplies of TOCOSOL Paclitaxel to Bayer Schering until such time as Bayer Schering establishes its own manufacturing capability.
On April 13, 2006, a wholly owned subsidiary of Bayer AG, a German corporation, or Bayer, submitted a formal tender offer to the stockholders of Schering AG to purchase all of the outstanding shares of Schering AG. The acquisition was essentially complete as of December 31, 2006 with only some minor activities remaining. We are not aware of any material effect the acquisition has had on our business, financial condition or results of operations but there can be no assurance that the acquisition will not have a material effect in the future.
3. Marketable Securities
Marketable securities consist of the following at December 31, 2006:
|
|
|
|
|
Unrealized |
|
Unrealized |
|
|
|
||||
|
|
|
Cost |
|
Gains |
|
Losses |
|
Fair Value |
|
||||
|
2006: |
|
|
|
|
|
|
|
|
|
||||
|
Corporate debt securities |
|
$ |
21,100,297 |
|
3,068 |
|
$ |
(1,111 |
) |
$ |
21,102,254 |
|
|
|
Mortgage-backed securities |
|
1,406,481 |
|
|
|
(2,649 |
) |
1,403,832 |
|
||||
|
|
|
$ |
22,506,778 |
|
$ |
3,068 |
|
$ |
(3,760 |
) |
$ |
22,506,086 |
|
There were no significant realized or unrealized gains or losses on the sales of marketable securities in 2006, 2005 or 2004. All of the marketable securities held as of December 31, 2006 had maturities of one year or less. The Company only invests in A (or equivalent) rated securities with maturities of one year or less. The Company does not believe that there are any permanent impairments related to unrealized losses for the year ended December 31, 2006 given the quality of the investment portfolio and its short-term nature. The Company held no marketable securities as of December 31, 2005.
4. Equipment, Furniture and Leasehold Improvements
Equipment, furniture and leasehold improvements consist of the following:
|
|
2006 |
|
2005 |
|
|||
|
Laboratory equipment |
|
$ |
3,973,654 |
|
$ |
3,595,440 |
|
|
Office furniture and equipment |
|
1,584,861 |
|
1,257,729 |
|
||
|
Leasehold improvements |
|
1,390,879 |
|
1,306,840 |
|
||
|
|
|
6,949,394 |
|
6,160,009 |
|
||
|
Less accumulated depreciation and amortization |
|
(5,763,220 |
) |
(5,153,606 |
) |
||
|
|
|
$ |
1,186,174 |
|
$ |
1,006,403 |
|
We held laboratory equipment acquired under capital leases with an original cost of $392,968 as of December 31, 2006 and 2005. Accumulated depreciation on this equipment was $380,500 and $355,600 at December 31, 2006 and 2005, respectively.
34
5. Accrued Expenses
Accrued expenses consist of the following:
|
|
2006 |
|
2005 |
|
|||
|
Clinical trials |
|
$ |
8,497,278 |
|
$ |
2,224,447 |
|
|
Product manufacturing |
|
1,617,580 |
|
438,332 |
|
||
|
Compensation |
|
1,459,128 |
|
1,455,329 |
|
||
|
Other |
|
354,138 |
|
289,736 |
|
||
|
|
|
$ |
11,928,124 |
|
$ |
4,407,844 |
|
6. Other assets
Other assets consist of the following:
|
|
2006 |
|
2005 |
|
|||
|
Deposit on facility lease |
|
$ |
439,822 |
|
$ |
51,500 |
|
|
Long-term portion of prepaid insurance |
|
20,895 |
|
52,239 |
|
||
|
|
|
$ |
460,717 |
|
$ |
103,739 |
|
7. Income Tax
The Company recorded no income tax expense or benefit during 2006, 2005 or 2004.
A reconciliation of the Federal Statutory tax rate of 34% to the Companys effective income tax rate follows:
|
|
2006 |
|
2005 |
|
2004 |
|
|
|
Statutory tax rate |
|
(34.00 |
%) |
(34.00 |
%) |
(34.00 |
%) |
|
Utilization of net operating loss carryforwards |
|
|
|
|
|
|
|
|
Permanent difference |
|
0.04 |
|
3.67 |
|
0.05 |
|
|
Change in valuation allowance |
|
35.08 |
|
33.63 |
|
33.95 |
|
|
Federal tax (refund) |
|
|
|
|
|
|
|
|
Other |
|
(1.12 |
) |
(3.30 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
Effective tax rate |
|
|
|
|
|
|
|
Significant components of the Companys net deferred tax assets and liabilities as of December 31, 2006 and 2005 are as follows:
|
|
2006 |
|
2005 |
|
|||
|
Deferred tax assets: |
|
|
|
|
|
||
|
Federal net operating loss carryforwards |
|
$ |
32,863,000 |
|
$ |
23,782,000 |
|
|
Deferred Revenue |
|
3,769,000 |
|
5,655,000 |
|
||
|
Accrued expenses |
|
195,000 |
|
166,000 |
|
||
|
Research and development credits |
|
3,119,000 |
|
2,651,000 |
|
||
|
Stock Options |
|
739,000 |
|
|
|
||
|
Book in excess of tax depreciation expense |
|
(62,000 |
) |
106,000 |
|
||
|
Gross deferred tax assets |
|
40,623,000 |
|
32,360,000 |
|
||
|
Valuation allowance for net deferred tax assets |
|
(40,623,000 |
) |
(32,360,000 |
) |
||
|
Net deferred tax assets |
|
$ |
|
|
$ |
|
|
Due to the uncertainty of the Companys ability to generate taxable income to realize its net deferred tax assets at December 31, 2006 and 2005, a valuation allowance has been recognized for financial reporting purposes. The Companys valuation allowance for deferred tax assets increased $8.3 million and $7.1 million for the years ended December 31, 2006 and 2005, respectively. The increase in the deferred tax assets in 2006 is primarily the result of increasing net operating loss carryforwards.
At December 31, 2006 the Company has federal net operating loss carryforwards of approximately $96.8 million for income tax reporting purposes and research and development tax credit carryforwards of approximately $3.1 million. The federal operating loss carryforwards and research and development credits will expire between 2007 and 2027. To the extent
35
that net operating loss carryforwards, when realized, relate to stock option deductions of approximately $2.8 million, the resulting benefit will be credited to stockholders equity.
The initial public offering of common stock by the Company in 1995 caused an ownership change pursuant to applicable regulations in effect under the Internal Revenue Code of 1986. Therefore, the Companys use of losses incurred through the date of ownership change will be limited during the carryforward period and may result in the expiration of net operating loss carryforwards before utilization.
8. Stockholders Equity
Common Stock
At December 31, 2006, the Company had shares of common stock reserved for possible future issuance as follows:
|
Stock options outstanding |
|
4,756,890 |
|
|
Warrants outstanding |
|
4,480,377 |
|
|
Shares available for future grant under stock plans |
|
1,051,011 |
|
|
|
|
10,288,278 |
|
Common Stock Issuances
In May 2006, the Company issued approximately 6.1 million shares of common stock in a registered direct offering for gross proceeds of $30.6 million (approximately $28.6 million net of transaction costs). The common stock was sold at a price of $5.00 per share and was previously registered through a shelf registration statement on Form S-3 that was declared effective by the SEC in April 2006.
In October 2005, the Company issued 3,900,000 shares of common stock and warrants to purchase 975,000 shares of common stock to Schering Berlin Venture Corporation for aggregate consideration of $15.8 million in connection with the Collaboration and License Agreement with Bayer Schering. The common stock was sold at $4.02 per share, which was equal to the per share closing price of the Companys common stock as reported on the Nasdaq National Market on October 14, 2005, the trading day immediately preceding the date of the Securities Purchase Agreement. The five-year warrants were sold at a price of $0.125 per share underlying each warrant, have an exercise price of $4.42 per share and expire in October 2010.
In August 2005, the Company sold 4.7 million shares of common stock and warrants to purchase up to 2.3 million shares of common stock in a private placement transaction for gross proceeds of $17.8 million (approximately $16.6 million net of transaction costs). The common stock was sold at a price of $3.77 per share. The five-year warrants were sold at a price of $0.125 per share underlying each warrant, have an exercise price of $4.15 per share and expire in August 2010.
Stock Warrants
At December 31, 2006, there were warrants outstanding to purchase 4.5 million shares of common stock at exercise prices ranging from $4.09 to $9.40 per share and expiration dates ranging from January 2007 to October 2010. During 2006, the Company recorded $301,330 in proceeds from the issuance of 73,675 shares of common stock from the exercise of common stock warrants. During 2005, the Company recorded $2,351,750 in proceeds from the issuance of 575,000 shares of common stock from the exercise of common stock warrants.
Stock Options
The Company has stock option plans whereby shares of common stock are reserved for future issuance pursuant to stock option grants or other issuances. Under the 2000 Stock Incentive Plan, an incremental number of shares equal to four percent of the Companys common stock outstanding as of December 31 of each year commencing December 31, 2000 are made available for issuance under the plan up to a lifetime maximum of five million shares. The Company reached the lifetime cap in 2006. Employee stock options vest over a period of time determined by the Board of Directors, generally four years, and director stock options are generally fully vested on the date of grant. Stock options generally are granted at the fair market value on the date of grant and expire ten years from the date of grant.
36
Adoption of SFAS 123R
The Company adopted the requirements of SFAS No. 123 (revised 2004), Share-Based Payment, (or SFAS 123R) on January 1, 2006, utilizing the modified prospective method. The Company uses the Black-Scholes-Merton option pricing model as the most appropriate fair-value method for its awards and recognizes compensation cost on a straight-line basis over its awards vesting periods in accordance with the provisions of SFAS 123R. In valuing its options using the Black-Scholes-Merton option pricing model, the Company makes assumptions about risk-free interest rates, dividend yields, volatility and weighted average expected lives, including estimated forfeiture rates, of the options. Risk-free interest rates are derived from United States treasury securities as of the option grant date. Dividend yields are based on the Companys historical dividend payments, which have been zero to date. Volatility is derived from the historical volatility of the Companys common stock as traded on NASDAQ. Forfeiture rates are estimated using historical actual forfeiture rates that resulted over the estimated life of the option grant for options granted as of the beginning of the forfeiture measurement period. These rates are adjusted on a quarterly basis and any change in compensation expense is recognized in the period of the change. The weighted average expected life of the options is based on historical experience of option exercises and the average vesting option schedule.
For unvested awards granted prior to the adoption date, compensation expense is based on the original grant date fair value measurement under SFAS 123, Accounting for Stock-Based Compensation. We currently believe that the assumptions used to generate those fair values are appropriate and therefore have not revised those calculations.
Prior to the adoption of SFAS 123R
The Company previously applied Accounting Principles Board (APB) Opinion No. 25, Accounting for Stock Issued to Employees, and related Interpretations and provided the required pro forma disclosures of SFAS No. 123.
The pro-forma information for the two annual periods ended December 31, 2005 was as follows:
|
|
2005 |
|
2004 |
|
|||
|
Net loss, as reported |
|
$ |
(21,097,017 |
) |
$ |
(16,310,592 |
) |
|
Add: Stock-based employee compensation expense included in reported net loss |
|
|
|
|
|
||
|
|
|
|
|
|
|
||
|
Deduct: Stock-based employee compensation expense determined under fair value based method |
|
(1,629,317 |
) |
(1,528,401 |
) |
||
|
|
|
|
|
|
|
||
|
Pro forma net loss |
|
$ |
(22,726,334 |
) |
$ |
(17,838,993 |
) |
|
|
|
|
|
|
|
||
|
Loss per share: |
|
|
|
|
|
||
|
Basic and diluted-as reported |
|
$ |
(0.88 |
) |
$ |
(0.81 |
) |
|
Basic and diluted-pro forma |
|
$ |
(0.95 |
) |
$ |
(0.88 |
) |
Impact of the adoption of SFAS 123R
The Company elected to implement SFAS 123R using the modified prospective application method. Accordingly, during the year ended December 31, 2006, the Company recorded stock-based compensation expense totaling the amount that would have been recognized had the fair value method been applied since the effective date of SFAS 123 for unvested options outstanding as of January 1, 2006 and recorded compensation expense under the provisions of SFAS 123R for options granted during the year ended December 31, 2006. Previously reported amounts have not been restated. As the Company uses a full valuation allowance with respect to deferred taxes, the adoption of SFAS 123R had no impact on deferred taxes or cash flow.
37
The effect of recording stock-based compensation for the year ended December 31, 2006 was as follows:
|
|
2006 |
|
||
|
|
|
|
|
|
|
Stock-based compensation expense: |
|
|
|
|
|
General & administrative |
|
$ |
(1,105,253 |
) |
|
Research & development |
|
(1,068,126 |
) |
|
|
Total stock-based compensation expense |
|
(2,173,379 |
) |
|
|
Tax effect on stock-based compensation |
|
|
|
|
|
Net effect on income |
|
$ |
(2,173,379 |
) |
|
|
|
|
|
|
|
Effect on earnings per share: |
|
|
|
|
|
Basic and diluted |
|
$ |
(0.06 |
) |
As of January 1, 2006, the Company had an unrecorded deferred stock-based compensation balance related to stock options of $5.4 million before estimated forfeitures. In the Companys pro forma disclosures prior to the adoption of SFAS 123R, the Company accounted for forfeitures upon occurrence. SFAS 123R requires forfeitures to be estimated at the time of grant and revised if necessary in subsequent periods if actual forfeitures differ from those estimates. Accordingly, as of January 1, 2006, the Company estimated that the stock-based compensation for the awards not expected to vest was $1.2 million, and therefore, the proforma deferred stock-based compensation balance related to stock options was adjusted to $4.2 million after estimated forfeitures.
As of December 31, 2006, the proforma deferred stock-based compensation balance related to stock options after adjusting for estimated forfeitures was $5.5 million and will be recognized over an estimated weighted average period of 2.7 years.
The fair value of each stock option used in the calculations under SFAS 123R is estimated using the Black-Scholes-Merton option pricing model. The assumptions used in this model include (1) the stock price at grant date, (2) the exercise price, (3) an estimated option life of four years, (4) no expected dividends for each period presented, (5) stock price volatility factor of 62.9%, 78.7% and 99.1% in 2006, 2005 and 2004, respectively, (6) forfeiture rate of 6.9% as of December 31, 2006, and (7) a risk-free interest rate of 4.6%, 4.4% and 3.5% in 2006, 2005 and 2004, respectively.
The Black-Scholes-Merton option pricing model was developed for use in estimating the fair value of short-lived exchange traded options that have no vesting restrictions and are fully transferable. In addition, option pricing models require the input of highly subjective assumptions, including the options expected life and the price volatility of the underlying stock. The Company will evaluate its assumptions on a regular basis. These evaluations may result in changes to assumptions which may have a material effect on compensation expense recorded under SFAS 123R.
A summary of activity related to the Companys stock options follows:
|
|
|
Shares |
|
Exercise |
|
||||||
|
Balance, December 31, 2003 |
|
2,588,652 |
|
$ |
0.63 |
|
|
|
$ |
44.00 |
|
|
Granted |
|
926,575 |
|
2.86 |
|
|
|
7.84 |
|
||
|
Exercised |
|
(136,670 |
) |
0.63 |
|
|
|
3.38 |
|
||
|
Canceled |
|
(368,048 |
) |
0.88 |
|
|
|
8.08 |
|
||
|
Balance, December 31, 2004 |
|
3,010,509 |
|
0.63 |
|
|
|
44.00 |
|
||
|
Granted |
|
1,039,000 |
|
2.87 |
|
|
|
5.10 |
|
||
|
Exercised |
|
(60,998 |
) |
0.88 |
|
|
|
4.06 |
|
||
|
Canceled |
|
(169,341 |
) |
2.03 |
|
|
|
8.19 |
|
||
|
Balance, December 31, 2005 |
|
3,819,170 |
|
0.63 |
|
|
|
44.00 |
|
||
|
Granted |
|
1,023,650 |
|
4.48 |
|
|
|
6.11 |
|
||
|
Exercised |
|
(53,720 |
) |
0.88 |
|
|
|
3.86 |
|
||
|
Canceled |
|
(32,210 |
) |
2.30 |
|
|
|
20.50 |
|
||
|
Balance, December 31, 2006 |
|
4,756,890 |
|
0.63 |
|
|
|
44.00 |
|
||
Options exercisable at December 31, 2006, 2005, and 2004, were 2,618,765, 1,953,680 and 1,485,380, respectively. The weighted average exercise prices for those options for the years ended December 31, 2006, 2005 and 2004, were $4.72, $4.83 and $4.13, respectively.
38
The intrinsic value of options exercised during 2006, 2005 and 2004 was $178,220, $109,644 and $437,280, respectively. The estimated fair value of shares vested during 2006, 2005 and 2004 was $2,310,842, $2,186,496 and $1,715,998, respectively. The weighted-average estimated fair value of stock options granted during 2006, 2005 and 2004 was $2.89, $2.99 and $3.09, respectively, based on the assumptions in the Black-Scholes-Merton valuation model discussed above.
The following table summarizes information about stock options outstanding at December 31, 2006:
|
|
|
Options Outstanding |
|
Options Exercisable |
|
||||||||||
|
Range of |
|
Number of |
|
Weighted- |
|
Weighted- |
|
Number of |
|
Weighted- |
|
Weighted- |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
$0.63 - $1.46 |
|
227,475 |
|
3.92 |
|
$ |
0.73 |
|
227,475 |
|
3.92 |
|
$ |
0.73 |
|
|
$1.46 - $3.72 |
|
1,057,947 |
|
7.07 |
|
$ |
2.85 |
|
774,126 |
|
6.77 |
|
$ |
2.75 |
|
|
$3.72 - $5.24 |
|
1,733,443 |
|
8.33 |
|
$ |
4.93 |
|
653,234 |
|
7.51 |
|
$ |
4.85 |
|
|
$5.24 - $6.94 |
|
1,529,738 |
|
6.98 |
|
$ |
6.17 |
|
759,733 |
|
4.22 |
|
$ |
6.27 |
|
|
$6.94 - $8.08 |
|
185,654 |
|
5.16 |
|
$ |
7.97 |
|
181,564 |
|
5.12 |
|
$ |
7.98 |
|
|
$8.08- $19.37 |
|
10,000 |
|
1.33 |
|
$ |
19.38 |
|
10,000 |
|
1.33 |
|
$ |
19.38 |
|
|
$19.37- $44.00 |
|
12,633 |
|
0.83 |
|
$ |
39.77 |
|
12,633 |
|
0.83 |
|
$ |
39.77 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
4,756,890 |
|
7.25 |
|
$ |
4.91 |
|
2,618,765 |
|
|
|
$ |
4.72 |
|
At December 31, 2006, the aggregate intrinsic value of the outstanding options was $6,848,112 and the aggregate intrinsic value of the exercisable options was $4,731,813.
Stock Purchase Plan
The Company has an employee stock purchase plan whereby employees may contribute up to 15% of their compensation to purchase shares of the Companys common stock at 85% of the stocks fair market value at the lower of the beginning or end of each six-month offering period. Shares purchased under the plan were 13,642, 6,493 and 3,390 in 2006, 2005 and 2004, respectively. At December 31, 2006, a total of 86,358 shares remain available for purchase by employees under the plan. The previous plan expired on December 31, 2005 and a new plan was approved by the shareholders at the 2006 annual meeting with a ten year term.
401(k) Plan
The Company has a 401(k) plan for all employees under which it provides a specified percentage match on employee contributions. Currently, the Company match is made in shares of the Companys common stock. Shares issued as matching contributions under the plan were 17,191, 18,591 and 10,568 in 2006, 2005 and 2004, respectively. The related expense recorded on these matching contributions was $92,762, $66,767 and $44,381 in 2006, 2005 and 2004, respectively. At December 31, 2006, a total of 11,287 shares remain available for future issuances as matching contributions under the plan.
Shareholder Rights Plan
The Company has adopted a Shareholder Rights Plan (Plan) which was amended in July 2002 and more recently in August 2006. Under the Plan, as amended, the Companys Board of Directors declared a dividend of one Preferred Stock Purchase Right (Right) for each outstanding common share of the Company. The Rights have an exercise price of $140 per Right and provide the holders with the right to purchase, in the event a person or group acquires 15% or more of the Companys common stock, additional shares of the Companys common stock having a market value equal to two times the exercise price of the Right. The Rights expire in 2016.
39
9. Net Loss Per Share
The following table presents the computation of basic and diluted net loss per share:
|
|
2006 |
|
2005 |
|
2004 |
|
||||
|
Basic and diluted net loss per share: |
|
|
|
|
|
|
|
|||
|
Net loss |
|
$ |
(23,551,296 |
) |
$ |
(21,097,017 |
) |
$ |
(16,310,592 |
) |
|
Weighted average common shares |
|
34,729,930 |
|
24,027,017 |
|
20,169,258 |
|
|||
|
Basic and diluted net loss per share |
|
$ |
(0.68 |
) |
$ |
(0.88 |
) |
$ |
(0.81 |
) |
As of December 31, 2006, 2005 and 2004 a total of 9,237,267, 8,373,222 and 4,838,625 options and warrants, respectively, have not been included in the calculation of potential common shares as their effect on diluted per share amounts would have been anti-dilutive.
10. Commitments and Contingencies
The Company has leased office space and equipment under three operating lease agreements, which expire in 2007. The Company signed a new facility lease in November 2006. The new facility lease has a term of 10 years with a provision for two additional five year renewals. The estimated commencement date for the new lease is October 2007. Future minimum lease payments under these leases are as follows:
|
2007 |
|
$ |
1,096,483 |
|
|
2008 |
|
1,762,681 |
|
|
|
2009 |
|
1,807,079 |
|
|
|
2010 |
|
1,852,809 |
|
|
|
2011 |
|
1,899,911 |
|
|
|
Thereafter |
|
11,906,566 |
|
|
|
|
|
$ |
20,325,529 |
|
Rental expense for the years ended December 31, 2006, 2005 and 2004 was $655,000, $644,000 and $647,000, respectively.
11. Quarterly Financial Information (unaudited)
|
|
Quarter Ended |
|
|||||||||||
|
|
|
Mar. 31 |
|
June 30 |
|
Sept. 30 |
|
Dec. 31 |
|
||||
|
|
|
(in thousands, except per share data) |
|
||||||||||
|
2006 |
|
|
|
|
|
|
|
|
|
||||
|
Collaboration revenue from Bayer Schering Pharma AG |
|
$ |
4,054 |
|
$ |
7,514 |
|
$ |
4,931 |
|
$ |
5,893 |
|
|
Operating expenses |
|
$ |
9,879 |
|
$ |
13,136 |
|
$ |
12,067 |
|
$ |
13,596 |
|
|
Operating loss |
|
$ |
(5,825 |
) |
$ |
(5,623 |
) |
$ |
(7,136 |
) |
$ |
(7,703 |
) |
|
Net loss |
|
$ |
(5,310 |
) |
$ |
(4,935 |
) |
$ |
(6,293 |
) |
$ |
(7,013 |
) |
|
Net loss per share*: |
|
|
|
|
|
|
|
|
|
||||
|
Basic and diluted |
|
$ |
(0.17 |
) |
$ |
(0.14 |
) |
$ |
(0.17 |
) |
$ |
(0.19 |
) |
|
|
|
|
|
|
|
|
|
|
|
||||
|
2005 |
|
|
|
|
|
|
|
|
|
||||
|
Collaboration revenue from Bayer Schering Pharma AG |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
8,254 |
|
|
Operating expenses |
|
$ |
4,866 |
|
$ |
4,165 |
|
$ |
9,042 |
|
$ |
11,991 |
|
|
Operating loss |
|
$ |
(4,866 |
) |
$ |
(4,165 |
) |
$ |
(9,042 |
) |
$ |
(3,736 |
) |
|
Net loss |
|
$ |
(4,775 |
) |
$ |
(4,091 |
) |
$ |
(8,908 |
) |
$ |
(3,323 |
) |
|
Net loss per share*: |
|
|
|
|
|
|
|
|
|
||||
|
Basic and diluted |
|
$ |
(0.22 |
) |
$ |
(0.19 |
) |
$ |
(0.37 |
) |
$ |
(0.11 |
) |
*Quarterly EPS may not add to annual figure due to rounding.
40
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15(e) under the Securities Exchange Act of 1934 (the Exchange Act)), as of the end of the period covered by this annual report on Form 10-K. Based on this evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of such date, our disclosure controls and procedures were effective.
In addition, no change in our internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) occurred during the fourth quarter of our fiscal year ended December 31, 2006 that has materially affected, or is reasonable likely to materially affect, our internal control over financial reporting.
41
Managements Report on Internal Control Over Financial Reporting
The management of Sonus Pharmaceuticals, Inc. (the Company) is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rule 13a15(f) under the Securities Exchange Act of 1934. The Companys internal control over financial reporting is a process designed under the supervision of the Companys principal executive and principal financial officers to provide reasonable assurance regarding the reliability of financial reporting and the preparation of the Companys financial statements for external reporting purposes in accordance with U.S. generally accepted accounting principles.
As of December 31, 2006, management assessed the effectiveness of the Companys internal control over financial reporting based on the framework established in Internal ControlIntegrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on this evaluation, management has determined that the Companys internal control over financial reporting was effective as of December 31, 2006.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Managements assessment of the effectiveness of its internal control over financial reporting as of December 31, 2006, has been audited by Ernst &Young LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
42
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Sonus Pharmaceuticals, Inc.
We have audited managements assessment, included in the accompanying Managements Report on Internal Control over Financial Reporting, that Sonus Pharmaceuticals, Inc. maintained effective internal control over financial reporting as of December 31, 2006, based on criteria established in Internal ControlIntegrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Sonus Pharmaceuticals, Inc.s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting. Our responsibility is to express an opinion on managements assessment and an opinion on the effectiveness of the companys internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, evaluating managements assessment, testing and evaluating the design and operating effectiveness of internal control, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A companys internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A companys internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the companys assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, managements assessment that Sonus Pharmaceuticals, Inc. maintained effective internal control over financial reporting as of December 31, 2006, is fairly stated, in all material respects, based on the COSO criteria. Also, in our opinion, Sonus Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2006, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of Sonus Pharmaceuticals, Inc. as of December 31, 2006 and December 31, 2005, and the related statements of operations, stockholders equity, and cash flows for each of the three years in the period ended December 31, 2006 of Sonus Pharmaceuticals, Inc., and our report dated March 13, 2007 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Seattle, Washington
March 13, 2007
43
ITEM 9B. OTHER INFORMATION
Not applicable.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
In compliance with Section 406 of the Sarbanes-Oxley Act of 2002 and the Nasdaq corporate governance listing standards, the Company has adopted a code of conduct that is applicable to all of the Companys employees and directors. Interested parties may request a copy of this code of conduct, free of charge, by delivering a written request addressed to the Chief Financial Officer, Sonus Pharmaceuticals, Inc., 22026 20th Avenue S.E., Bothell, Washington 98021. The Company will disclose any amendments to the code of conduct and any waivers from the code of conduct for directors and executive officers by posting such information on its website at www.sonuspharma.com.
The information required hereunder is incorporated by reference from our Proxy Statement to be filed within 120 days of December 31, 2006 and delivered to stockholders in connection with our 2007 Annual Meeting of Stockholders to be held May 10, 2007.
ITEM 11. EXECUTIVE COMPENSATION
The information required hereunder is incorporated by reference from our Proxy Statement to be filed within 120 days of December 31, 2006 and delivered to stockholders in connection with our 2007 Annual Meeting of Stockholders to be held May 10, 2007.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information regarding our equity compensation plans as of December 31, 2006:
|
|
|
(a) |
|
(b) |
|
(c) |
|
|
|
Plan category |
|
Number of securities |
|
Weighted-average |
|
Number of securities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity compensation plans approved by security holders (1) |
|
4,272,682 |
|
$ |
4.88 |
|
1,036,793 |
|
|
Equity compensation plans not approved by security holders (2) |
|
484,208 |
|
$ |
5.05 |
|
2,931 |
|
|
Total |
|
4,756,890 |
|
|
|
1,039,724 |
|
|
(1) Our 2000 Stock Incentive Plan was approved by security holders with 500,000 shares authorized under the plan. Stock options issued under the 2000 plan are generally granted at the fair market value on the date of grant and expire ten years from the date of grant. The plan also has an annual feature whereby an incremental number of shares equal to four percent of the Companys common stock outstanding as of December 31 of each year commencing December 31, 2000 are made available for issuance under the plan up to a lifetime maximum of five million shares. This lifetime maximum was reached in 2006. 950,435 shares were available for issuance as of December 31, 2006. The Company also had 86,358 shares available at December 31, 2006 for issuance under its Employee Stock Purchase Plan. This plan expires in 2016.
(2) Our 1999 Nonqualified Stock Incentive Plan (the 1999 Plan) is a broad-based plan for which shareholder approval was not required or obtained. A total of 900,000 shares are authorized under the 1999 Plan with 2,931 available for issuance as of December 31, 2006. Options to purchase 484,208 shares of common stock under the 1999 Plan were outstanding as of December 31, 2006 at a weighted average exercise price of $5.05. Stock options issued under the 1999 Plan are generally granted with an exercise price equal to fair market value on the date of grant, but in no event may be less than 85% of the then fair market value. Options under the 1999 Plan have various vesting schedules and expire ten years from the date of grant. The 1999 Plan also authorizes the issuance of restricted stock, although no restricted stock grants have been issued under the 1999 Plan. Shares underlying unexercised options that expire or are terminated become available again for future grants.
44
The remaining information required hereunder is incorporated by reference from our Proxy Statement to be filed within 120 days of December 31, 2006 and delivered to stockholders in connection with its 2007 Annual Meeting of Stockholders to be held May 10, 2007.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required hereunder is incorporated by reference from our Proxy Statement to be filed within 120 days of December 31, 2006 and delivered to stockholders in connection with our 2007 Annual Meeting of Stockholders to be held May 10, 2007.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required hereunder is incorporated by reference from our Proxy Statement to be filed within 120 days of December 31, 2006 and delivered to stockholders in connection with its 2007 Annual Meeting of Stockholders to be held May 10, 2007.
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
|
(a) |
(1) Financial Statements |
|
|
|
|
|
|
|
|
|
Report of Independent Registered Public Accounting Firm |
26 |
|
|
|
|
|
|
|
|
Balance Sheets as of December 31, 2006 and 2005 |
27 |
|
|
|
|
|
|
|
|
Statements of Operations for the years ended December 31, 2006, 2005, and 2004 |
28 |
|
|
|
|
|
|
|
|
Statements of Stockholders Equity for the years ended December 31, 2006, 2005, and 2004 |
29 |
|
|
|
|
|
|
|
|
Statements of Cash Flows for the years ended December 31, 2006, 2005, and 2004 |
30 |
|
|
|
|
|
|
|
|
Notes to the Financial Statements |
31 |
|
|
|
|
|
|
|
|
(2) All schedules are omitted because they are not required or the required information is included in the financial statements or notes thereto. |
|
|
|
|
|
|
|
|
|
(3) Exhibits |
|
|
|
Exhibit No. |
|
Index to Exhibits |
|
Location |
|
|
|
|
|
|
|
|
|
|
Exhibit No. 2: Plan of Acquisition |
|
|
||
|
|
2.1 |
|
Stock Purchase Agreement, dated November 3, 2004 |
|
(22) |
|
|
2.2 |
|
Amended and Restated Stock Purchase Agreement, dated December 22, 2004. |
|
(23) |
|
|
|
|
|
|
|
|
|
Exhibit No. 3: Articles of Incorporation |
|
|
||
|
|
3.2 |
|
Amended and Restated Certificate of Incorporation of the Company. |
|
(1) |
|
|
3.3 |
|
Certificate of Amendment of Certificate of Incorporation of the Company. |
|
(7) |
|
|
3.4 |
|
Amended and Restated Bylaws of the Company. |
|
(1) |
|
|
3.5 |
|
Amended and Restated Certificate of Incorporation of the Company. |
|
(20) |
|
|
|
|
|
|
|
|
|
Exhibit No. 4: Instruments Defining the Rights of Security Holders |
|
|
||
|
|
4.1 |
|
Specimen Certificate of Common Stock. |
|
(1) |
|
|
4.2 |
|
Rights Agreement, dated as of August 23, 1996, between the Company and U.S. Stock Transfer Corporation. |
|
(3) |
|
|
4.3 |
|
First Amendment to Rights Agreement, dated as of August 23, 1996, between the Company and U.S. Stock Transfer Corporation. |
|
(17) |
|
|
4.4 |
|
Second Amendment to Amended and Restated Rights Agreement, dated August 10, 2006, between the Company and U.S. Stock Transfer Corporation, as Rights Agent. |
|
(29) |
45
|
Exhibit No. 10:
Material Contracts |
|
|
|||
|
|
Compensation Plans and Arrangements |
|
|
||
|
|
10.1 |
|
Sonus Pharmaceuticals, Inc. Incentive Stock Option, Nonqualified Stock Option and Restricted Stock Purchase Plan 1991 (the 1991 Plan), as amended. |
|
(1) |
|
|
10.2 |
|
Form of Incentive Stock Option Agreement pertaining to the 1991 Plan. |
|
(1) |
|
|
10.3 |
|
Form of Nonqualified Stock Option Agreement pertaining to the 1991 Plan. |
|
(1) |
|
|
10.4 |
|
Form of Restricted Stock Purchase Agreement pertaining to the 1991 Plan. |
|
(1) |
|
|
10.5 |
|
Sonus Pharmaceuticals, Inc. 1995 Stock Option Plan for Directors (the Director Plan). |
|
(1) |
|
|
10.6 |
|
Form of Stock Option Agreement pertaining to the Director Plan. |
|
(1) |
|
|
10.7 |
|
1999 Nonqualified Stock Incentive Plan (the 1999 Plan). |
|
(7) |
|
|
10.8 |
|
Form of Stock Option Agreement pertaining to the 1999 Plan. |
|
(7) |
|
|
10.9 |
|
Form of Restricted Stock Purchase Agreement pertaining to the 1999 Plan. |
|
(7) |
|
|
10.10 |
|
2000 Stock Incentive Plan (the 2000 Plan). |
|
(9) |
|
|
10.11 |
|
Form of Stock Option Agreement pertaining to the 2000 Plan. |
|
(9) |
|
|
10.12 |
|
Sonus Pharmaceuticals, Inc. Employee Stock Purchase Plan. |
|
(2) |
|
|
10.13 |
|
Change in Control Agreement for Michael Martino, dated September 15, 1998. |
|
(4) |
|
|
10.14 |
|
Change in Control Agreement for Richard J. Klein, dated October 25, 2000. |
|
(10) |
|
|
10.15 |
|
Change in Control Agreement for Michael A. Martino, dated July 18, 2001. |
|
(12) |
|
|
10.16 |
|
Change in Control Agreement for Michael B. Stewart, dated May 1, 2003. |
|
(18) |
|
|
10.17 |
|
Change in Control Agreement for Michael A. Martino, dated October 10, 2003. |
|
(19) |
|
|
10.18 |
|
Change in Control Agreement for Richard J. Klein, dated October 10, 2003. |
|
(19) |
|
|
10.19 |
|
Change in Control Agreement for Michael B. Stewart, dated October 10, 2003. |
|
(19) |
|
|
10.20 |
|
Change in Control Agreement for Alan Fuhrman, dated September 15, 2004. |
|
(22) |
|
|
10.21 |
|
Amended and Restated Executive Compensation Program. |
|
(25) |
|
|
10.22 |
|
Form of Performance Award under Executive Compensation Program. |
|
(25) |
|
|
10.23 |
|
First Amendment to Sonus Pharmaceuticals, Inc. 2000 Stock Incentive Plan |
|
(30) |
|
|
10.24 |
|
Sonus Pharmaceuticals, Inc. Compensation Policy |
|
(31) |
|
|
10.25 |
|
Sonus Pharmaceuticals, Inc. Executive Compensation Program |
|
(31) |
|
|
|
|
|
|
|
|
|
Other Material Contracts |
|
|
||
|
|
10.20 |
|
Lease Agreement dated January 17, 1994 between the Company and WRC Properties, Inc. |
|
(1) |
|
|
10.21 |
|
Amendment 2 dated October 28, 1997 to Lease Agreement dated January 17, 1994. |
|
(5) |
|
|
10.22 |
|
Amendment 3 dated October 15, 1998 to Lease Agreement dated January 17, 1994. |
|
(5) |
|
|
10.23 |
|
Amendment 4 dated November 29, 2001 to Lease Agreement dated January 17, 1994. |
|
(15) |
|
|
10.24 |
|
Form of Indemnification Agreement for Officers and Directors of the Company. |
|
(1) |
|
|
10.25 |
|
License Agreement by and between Nycomed Amersham AS and the Company dated August 31, 1999. |
|
(8) |
|
|
10.26 |
|
License Agreement by and between Chugai Pharmaceutical Co. Ltd., Molecular Biosystems, Inc., and the Company, dated December 22, 2000. |
|
(11) |
|
|
10.27 |
|
Nycomed Assignment and Asset Transfer Agreement, dated August 3, 2001. |
|
(13) |
|
|
10.28 |
|
Supply Agreement dated January 22, 2002 between Indena SpA and Sonus Pharmaceuticals, Inc. |
|
(14) |
|
|
10.29 |
|
First Amendment dated May 5, 2003 to Supply Agreement dated January 22, 2002 between Indena SpA and Sonus Pharmaceuticals, Inc. |
|
(18) |
|
|
10.30 |
|
Manufacturing and Supply Agreement by and between the Company and Gensia Sicor Pharmaceutical Sales, Inc., dated June 26, 2002. |
|
(16) |
|
|
10.31 |
|
Securities Purchase Agreement, dated May 7, 2004. |
|
(21) |
|
|
10.32 |
|
Registration Rights Agreement, dated May 7, 2004. |
|
(21) |
|
|
10.33 |
|
Securities Purchase Agreement, dated August 15, 2005. |
|
(26) |
46
|
10.34 |
|
Registration Rights Agreement, dated August 15, 2005. |
|
(26) |
|
|
|
10.35 |
|
Collaboration and License Agreement by and between the Company and Bayer Schering Pharma AG, dated October 17, 2005. |
|
(27) |
|
|
10.36 |
|
Securities Purchase Agreement, dated October 17, 2005. |
|
(27) |
|
|
10.37 |
|
Registration Rights Agreement, dated October 17, 2005. |
|
(27) |
|
|
10.38 |
|
Form of Purchase Agreement, dated April 27, 2006 |
|
(28) |
|
|
10.39 |
|
Lease Agreement dated November 21, 2006 between the Company and BMR-217TH Place LLC |
|
(6) |
|
|
10.40 |
|
Clinical Supply Agreement between the Company and Bayer Schering Pharma AG |
|
(6) |
|
|
|
|
|
|
|
|
|
Exhibit No. 23: Consents of Experts and Counsel |
|
|
||
|
|
23.1 |
|
Consent of Independent Registered Public Accounting Firm. |
|
(6) |
|
|
24.1 |
|
Power of Attorney (included on the Signature Page of this Annual Report on Form 10-K). |
|
(6) |
|
|
|
|
|
|
|
|
|
Certifications |
|
|
||
|
|
31.1 |
|
Certification of President and Chief Executive Officer pursuant to Rule 13a-14(a) or 15d-14(a). |
|
(6) |
|
|
31.2 |
|
Certification of the Chief Financial Officer pursuant to Rule 13a-14(a) or 15d-14(a). |
|
(6) |
|
|
32.1 |
|
Certification of President and Chief Executive Officer pursuant to Rule 13a-14(b) or 15d-14(b). |
|
(6) |
|
|
32.2 |
|
Certification of the Chief Financial Officer pursuant to Rule 13a-14(b) or 15d-14(b). |
|
(6) |
(1) Incorporated by reference to the Companys Registration Statement on Form S-1, Reg. No. 33-96112.
(2) Incorporated by reference to the Companys Registration Statement on Form S-1, Reg. No. 33-80623.
(3) Incorporated by reference to the Companys Registration Statement on Form 8-A, dated August 23, 1996.
(4) Incorporated by, reference to the Companys Quarterly Report on Form 10-Q for the quarterly period ended September 30, 1998.
(5) Incorporated by reference to the Companys Annual Report on Form 10-K for the period ended December 31, 1998.
(6) Filed herewith.
(7) Incorporated by reference to the Companys Quarterly Report on Form 10-Q for the quarterly period ended March 31, 1999.
(8) Incorporated by reference to the Companys Current Report on Form 8-K dated September 28, 1999.
(9) Incorporated by reference to the Companys Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2000.
(10) Incorporated by reference to the Companys Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2000.
(11) Incorporated by reference to the Companys Annual Report on Form 10-KA for the period ended December 31, 2000.
(12) Incorporated by reference to the Companys Quarterly Report on Form 10-QA for the quarterly period ended June 30, 2001.
(13) Incorporated by reference to the Companys Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2001.
(14) Incorporated by reference to the Companys Registration Statement on Form S-3 filed February 8, 2002.
(15) Incorporated by reference to the Companys Annual Report on Form 10-K for the period ended December 31, 2001.
(16) Incorporated by reference to the Companys Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2002.
(17) Incorporated by reference to the Companys filing on Form 8-A12G/A dated July 25, 2002.
(18) Incorporated by reference to the Companys Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2003.
47
(19) Incorporated by reference to the Companys Annual Report on Form 10-K for the period ended December 31, 2003.
(20) Incorporated by reference to the Companys Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2004.
(21) Incorporated by reference to the Companys Current Report on Form 8-K filed May 13, 2004.
(22) Incorporated by reference to the Companys Current Report on Form 8-K filed September 20, 2004.
(23) Incorporated by reference to the Companys Current Report on Form 8-K filed November 8, 2004.
(24) Incorporated by reference to the Companys Current Report on Form 8-K filed December 28, 2004.
(25) Incorporated by reference to the Companys Current Report on Form 8-K filed January 4, 2005.
(26) Incorporated by reference to the Companys Current Report on Form 8-K filed August 17, 2005.
(27) Incorporated by reference to the Companys Annual Report on Form 10-K for the period ended December 31, 2005.
(28) Incorporated by reference to the Companys Current Report on Form 8-K filed April 28, 2006.
(29) Incorporated by reference to the Companys Current Report on Form 8-K-A/A filed August 10, 2006.
(30) Incorporated by reference to the Companys Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2006.
(31) Incorporated by reference to the Companys Current Report on Form 8-K filed December 15, 2006.
48
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized, in the City of Bothell, State of Washington, on March 16, 2007.
|
|
SONUS PHARMACEUTICALS, INC. |
|
||
|
|
|
|
|
|
|
Dated: March 16, 2007 |
|
By: |
/s/ Michael A. Martino |
|
|
|
|
Michael A. Martino |
|
|
|
|
|
President, Chief Executive Officer |
|
|
We, the undersigned directors and officers of Sonus Pharmaceuticals, Inc., do hereby constitute and appoint Michael A. Martino and Alan Fuhrman, or either of them, our true and lawful attorneys and agents, with full powers of substitution to do any and all acts and things in our name and on behalf in our capacities as directors and officers and to execute any and all instruments for us and in our names in the capacities indicated below, which said attorneys and agents may deem necessary or advisable to enable said corporation to comply with the Securities Exchange Act of 1934, as amended, and any rules, regulations and requirements of the Securities and Exchange Commission, in connection with this Annual Report on Form 10-K, including specifically but without limitation, power and authority to sign for us or any of us in our names in the capacities indicated below, any and all amendments thereto; and we do hereby ratify and confirm all that said attorneys and agents, shall do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
/s/ Michael A. Martino |
|
President, Chief Executive |
|
|
|
March 16, 2007 |
|
Michael A. Martino |
|
Officer and Director (Principal Executive Officer) |
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Alan Fuhrman |
|
Senior Vice President, |
|
|
|
March 16, 2007 |
|
Alan Fuhrman |
|
Chief Financial Officer (Principal Financial Officer) |
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Craig S. Eudy |
|
Vice President, |
|
|
|
March 16, 2007 |
|
Craig S. Eudy |
|
Corporate Controller (Principal Accounting Officer) |
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Michelle Burris |
|
Director |
|
|
|
March 16, 2007 |
|
Michelle Burris |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ George W. Dunbar, Jr. |
|
Director |
|
|
|
March 16, 2007 |
|
George W. Dunbar, Jr. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Robert E. Ivy |
|
Director, Chairman of |
|
|
|
March 16, 2007 |
|
Robert E. Ivy |
|
the Board of Directors |
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Dwight Winstead |
|
Director |
|
|
|
March 16, 2007 |
|
Dwight Winstead |
|
|
|
|
|
|
49